基于UPLC特征图谱及主要成分含量测定的百合药材初加工方法研究
2021-08-26陈江平徐东婷程学仁
张 正,陈江平,钟 霞,徐东婷,胡 懿,程学仁
(广东一方制药有限公司 广东省中药配方颗粒企业重点实验室,广东 佛山 528244)
百合为百合科植物卷丹LiliumlancifoliumThumb.、百合LiliumbrowniiF.E.Brown var.viridulumBaker,或细叶百合LiliumpumilumDC.的干燥肉质鳞叶,具有养阴润肺、清心安神的功效,用于阴虚燥咳、劳嗽咳血、虚烦惊悸、失眠多梦、精神恍惚等证[1]。百合始载于《神农本草经》,被列为中品,为常用中药材,也是原国家卫生部2002年确定的药食同源品种之一[2-4]。研究[5-9]表明,百合中主要含有皂苷类、酚性甘油酯类、多糖类、磷脂类、生物碱类等活性成分,具有止咳祛痰、镇静催眠、免疫调节、抗肿瘤、抗应激损伤、抗氧化等药理作用,其中,王百合苷B为百合酚性甘油酯类成分之一[6],王百合苷B在百合药材中相对百分含量较高,水溶性好,为百合基础物质的主要成分之一。2015年版《中华人民共和国药典》百合项下仅有性状、薄层鉴别及浸出物的质量要求,本研究旨在建立百合药材王百合苷B含量测定方法及UPLC特征图谱方法,为百合药材的质量控制提供可靠依据。
初加工是影响中药材质量的关键环节,2015年版《中华人民共和国药典》规定百合鲜品的初加工方法为“秋季采挖,洗净,剥取鳞叶,置沸水中略烫,干燥”,笔者于安徽、湖南、浙江等百合主产区走访调研发现,百合的初加工方法除烫制法外,还有蒸制法、冷水浸泡法及直接干燥法,其初加工方法无规范化技术指南,这是导致百合药材质量差异的重要因素[10-13]。本研究以外观性状、浸出物、王百合苷B含量及UPLC特征图谱为评价指标,比较直接烘干法、冷水浸泡法、烫制法及蒸制法对百合药材质量的影响,为百合初加工的规范化操作提供依据。
1 仪器与试药
1.1 仪器
Waters e2695高效液相色谱仪(美国Waters公司);Waters H-Class高效液相色谱仪(美国Waters公司);ME204E型万分之一天平、XP26型百万分之一天平(瑞士梅特勒-托利多公司);KQ-500DE型数控超声波清洗器(昆山市超声仪器有限公司);Milli-Q Direct超纯水系统(默克股份有限公司)。
1.2 试剂与药物
王百合苷B对照品(批号:114420-66-5,南京世洲生物技术有限公司,纯度>98%);磷酸和乙腈为色谱纯;水为超纯水;其他试剂均为分析纯。
18批百合药材的产地信息见表1,产地加工方法各异,洗净剥片,开水烫或蒸,以鳞片边缘柔软而中部未熟,背面有极小的裂纹为度,无统一方法。鲜百合样品于2019年8月采自湖南省湘西土家族苗族自治州龙山县,所有样品经广东一方制药有限公司魏梅主任药师鉴定为正品,基原为卷丹,样品存放于广东一方制药有限公司。

表1 18批百合药材产地信息
2 评价指标测定方法
2.1 水分测定
按照水分测定法(2015年版《中华人民共和国药典》[14]四部通则0832)项下的第二法测定。
2.2 浸出物测定
按照浸出物测定法(2015年版《中华人民共和国药典》[14]四部通则2201)项下水溶性浸出物测定法的冷浸法和醇溶性浸出物测定法的热浸法测定。
2.3 王百合苷B含量测定
2.3.1 色谱条件 Waters XBridge C18色谱柱(4.6 mm×250 mm,5 μm),流动相乙腈-0.1%磷酸溶液(19∶81),流速1.0 mL·min-1,柱温30 ℃,进样量10 μL,检测波长312 nm。色谱见图1。
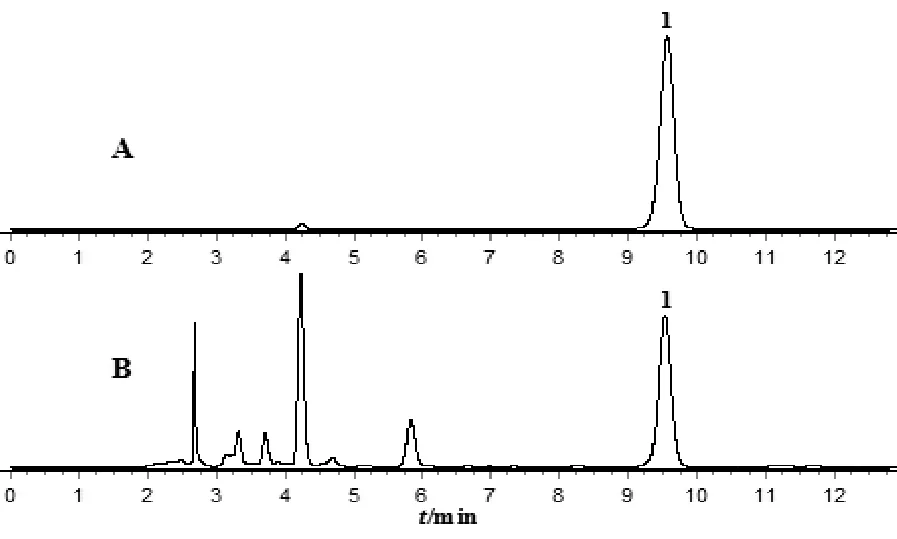
注:A.对照品溶液;B.供试品溶液;1.王百合苷B。
2.3.2 对照品溶液制备 取王百合苷B对照品适量,精密称定,置于10 mL量瓶中,加甲醇制成每1 mL含王百合苷B 292.352 μg的溶液,即得。
LAS方法和本文方法的迭代过程如表5和表6所示。可以看出,由于考虑了克里金近似的误差,本文方法的局域半径稍大于LAS方法,采样效率更高,求解精度也更好。
2.3.3 供试品溶液制备 取百合药材粉末(过三号筛)约2 g,精密称定,置具塞锥形瓶中,加入稀乙醇25 mL,称定重量,超声处理(功率300 W,频率45 kHz)30 min,放冷,再称定重量,用稀乙醇补足损失重量,摇匀,滤过,取续滤液,即得。
2.3.4 线性关系考察 取浓度为292.352 μg·mL-1的王百合苷B对照品溶液1 mL,分别置于1、2、5、10、25、50 mL量瓶中,加甲醇定容至刻度,按“2.3.1”项下色谱条件测定,以峰面积为纵坐标,对照品浓度为横坐标,得王百合苷B回归方程:Y=29 985.878 3X-107 432.617 7(r=0.999 1),线性范围为5.847~292.352 μg·mL-1。
2.3.5 精密度试验 精密吸取王百合苷B的对照品溶液,按“2.3.1”项下色谱条件重复进样6次,王百合苷B的峰面积RSD为2.52%,说明仪器精密度良好。
2.3.6 重复性试验 取百合药材样品粉末(S7),称取6份,按“2.3.3”项下方法制备供试品溶液,按“2.3.1”项下色谱条件测定,计算王百合苷B含量的RSD为0.93%,说明该方法的重复性良好。
2.3.7 稳定性试验 取百合药材样品(S7),按“2.3.3”项下方法制备供试品溶液,分别于制备后0、2、4、8、12、24 h按“2.3.1”项下色谱条件测定,计算王百合苷B含量的RSD为0.30%,说明供试品溶液在24 h内稳定性良好。
2.3.8 加样回收试验 取已知含有量的百合药材样品(S7)粉末适量,精密称取9份,每份约1.0 g,置于具塞锥形瓶中,分为3组,每组分别精密加入相当于含有量50%、100%、150%的王百合苷B对照品溶液,按“2.3.3”项下方法制备供试品溶液,按“2.3.1”项下色谱条件测定,计算平均加样回收率为103.84%,RSD为1.6%,说明该方法准确度良好。见表2。
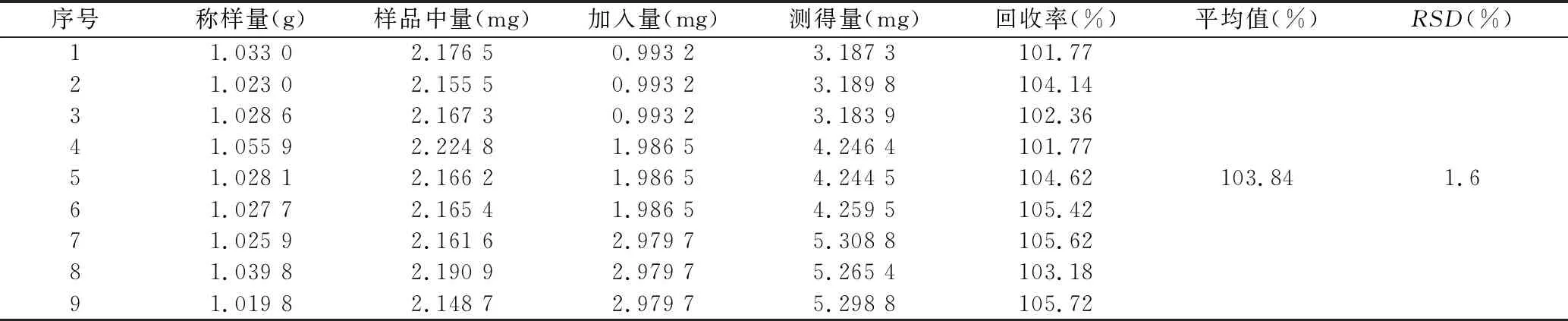
表2 王百合苷B加样回收实验 (n=9)
2.4 UPLC特征图谱方法建立
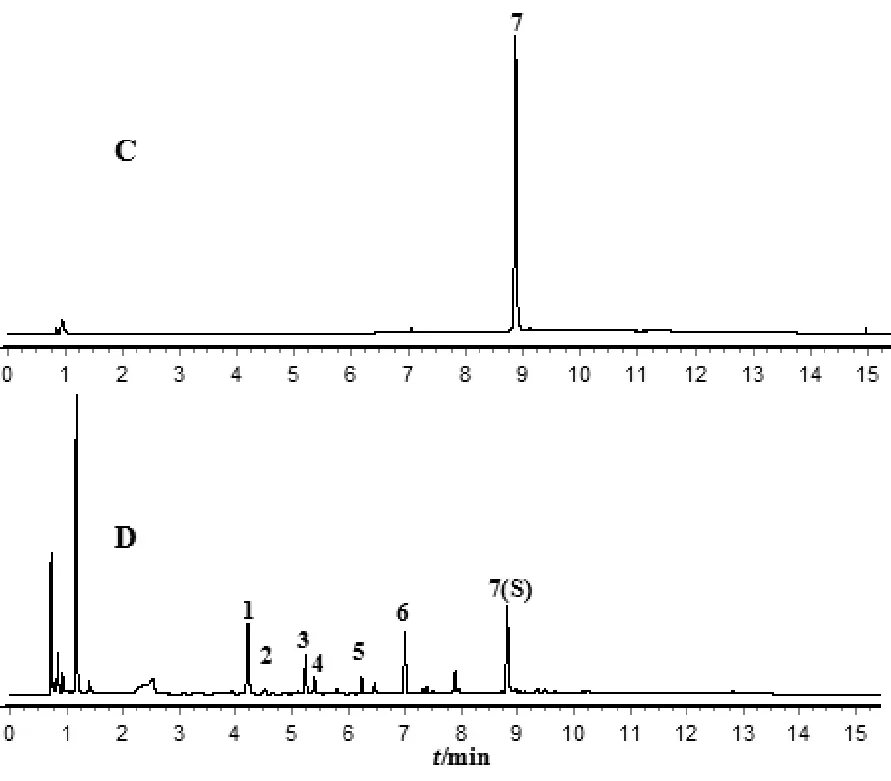
注:C.对照品溶液;D.供试品溶液;7.王百合苷B。
2.4.2 对照品溶液制备 同“2.3.2”项。
2.4.3 供试品溶液制备 同“2.3.3”项。
2.4.4 精密度试验 取“2.4.3”项下S3供试品溶液,按“2.4.1”项下色谱条件连续进样6次,以7号峰王百合苷B为参照峰,计算各色谱峰相对保留时间的RSD在0.05%~0.11%,相对峰面积的RSD为0.41%~1.89%,说明仪器精密度良好。
2.4.5 重复性试验 取同一批百合药材S3,按“2.4.3”项下方法平行制备6份供试品溶液,按“2.4.1”项下色谱条件测定,以7号峰王百合苷B为参照峰,计算各色谱峰相对保留时间的RSD在0.04%~0.10%,相对峰面积的RSD为1.41%~2.18%,说明该方法重复性良好。
2.4.6 稳定性试验 取“2.4.3”项下S3供试品溶液,分别于制备后0、2、4、8、12、24 h按“2.4.1”项下色谱条件测定,以7号峰王百合苷B为参照峰,计算各色谱峰相对保留时间的RSD为0.08%~0.14%,相对峰面积的RSD为1.95%~2.52%,说明供试品溶液在24 h内稳定。
2.4.7 特征图谱建立及色谱峰归属 取百合药材S1-S18,按“2.4.3”项下方法制备供试品溶液,按“2.4.1”项下色谱条件测定,记录色谱图,采用“中药色谱指纹图谱相似度评价系统”(2012版)进行分析。结果发现,18批百合药材共有7个分离较好的特征峰,其UPLC特征图谱见图3。通过与对照品色谱图对照,确定7号峰为王百合苷B,见图4。以7号峰王百合苷B为参照峰,计算其他特征峰的相对保留时间及相对峰面积,见表3、表4。

图3 18批百合药材UPLC特征图谱叠加
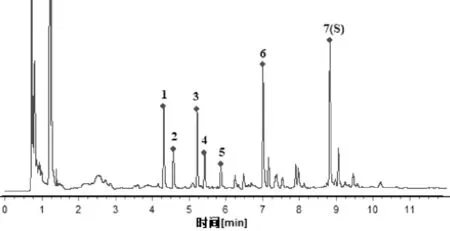
注:7.王百合苷B。

表3 18批百合药材共有峰相对保留时间
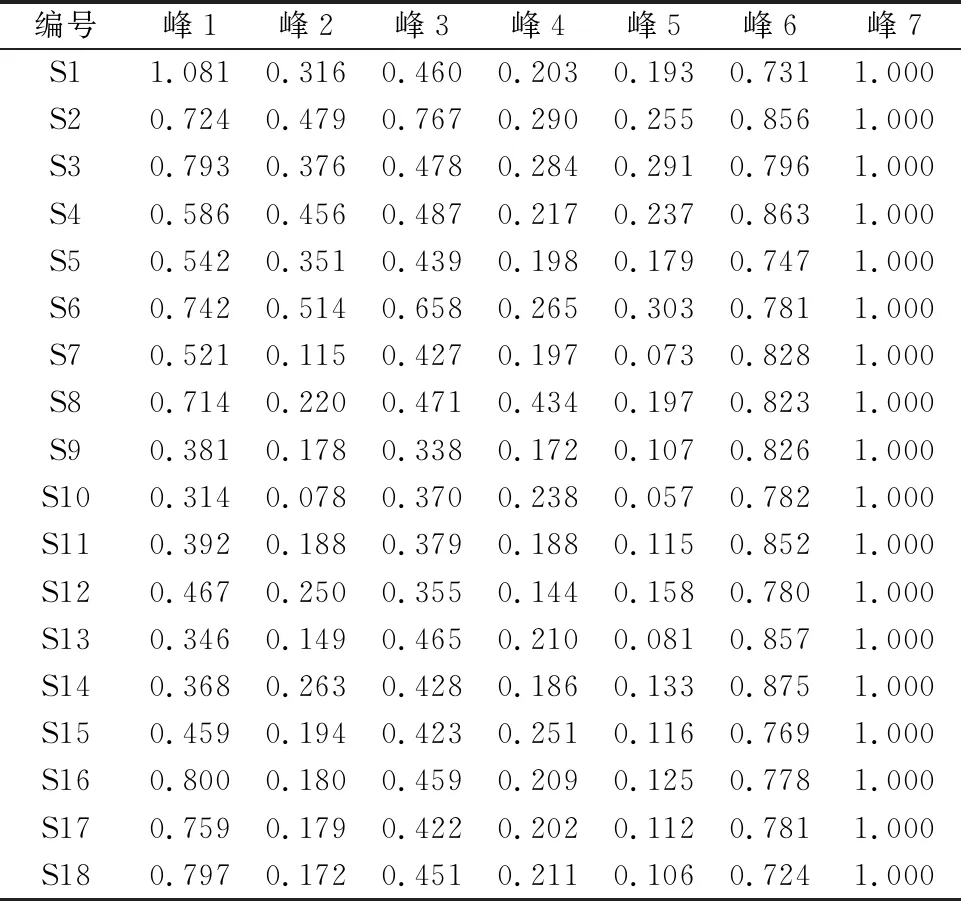
表4 18批百合药材共有峰相对峰面积
3 研究内容与结果
3.1 样品制备
取鲜百合样品,进行剥片、清洗、沥水、混匀,再进行不同初加工方法处理。见表5。
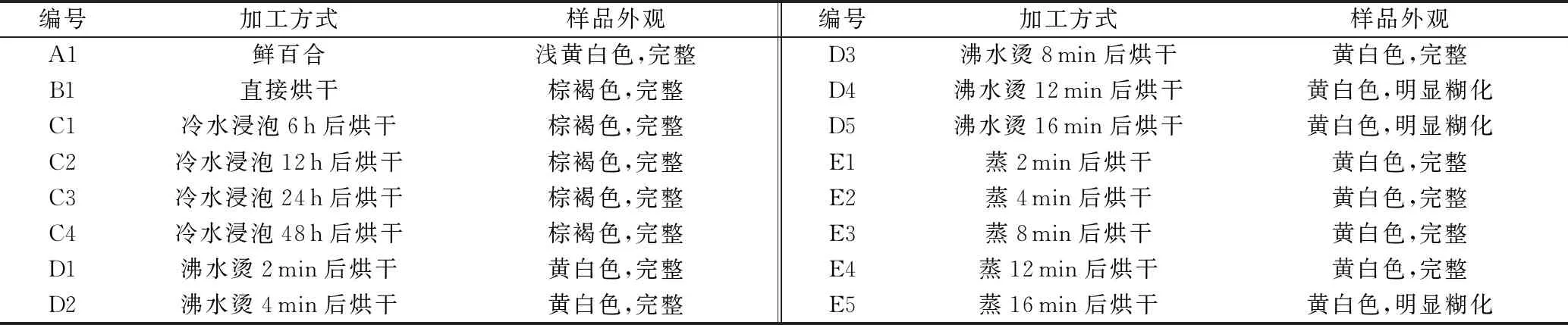
表5 百合样品加工方式与性状
3.2 百合样品水分与浸出物测定
取“3.1”项下百合样品,按“2.1”及“2.2”项下方法测定,结果见表6。

表6 百合样品水分与浸出物测定结果 (n=3)
3.3 含量测定
取“3.1”项下百合样品,按“2.3”项下方法测定,结果见表7。

表7 百合样品王百合苷B含量测定结果 (n=3)
3.4 特征图谱测定
取“3.1”项下样品,按“2.4”项下方法测定,各样品的相对保留时间与相对峰面积见表8、表9,UPLC特征图谱见图5。
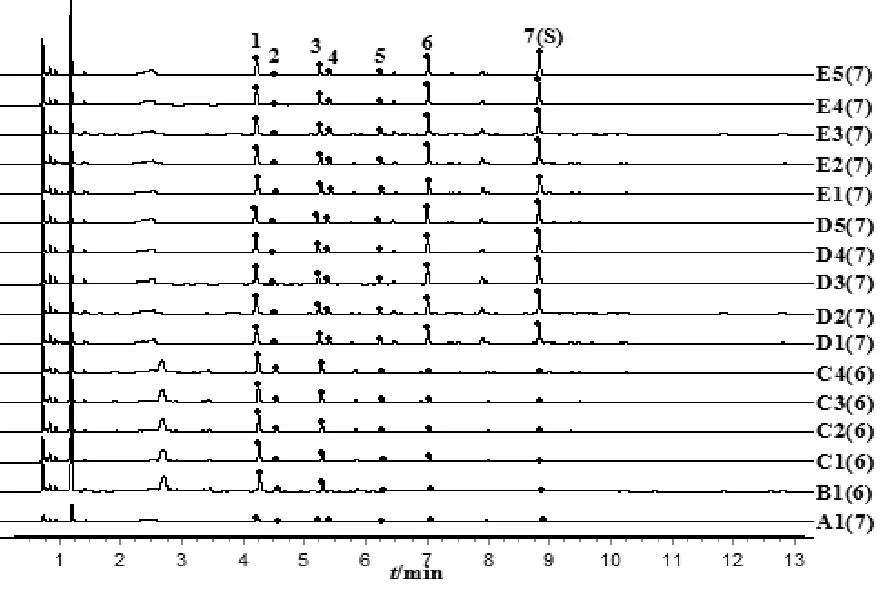
图5 百合样品UPLC特征图谱叠加
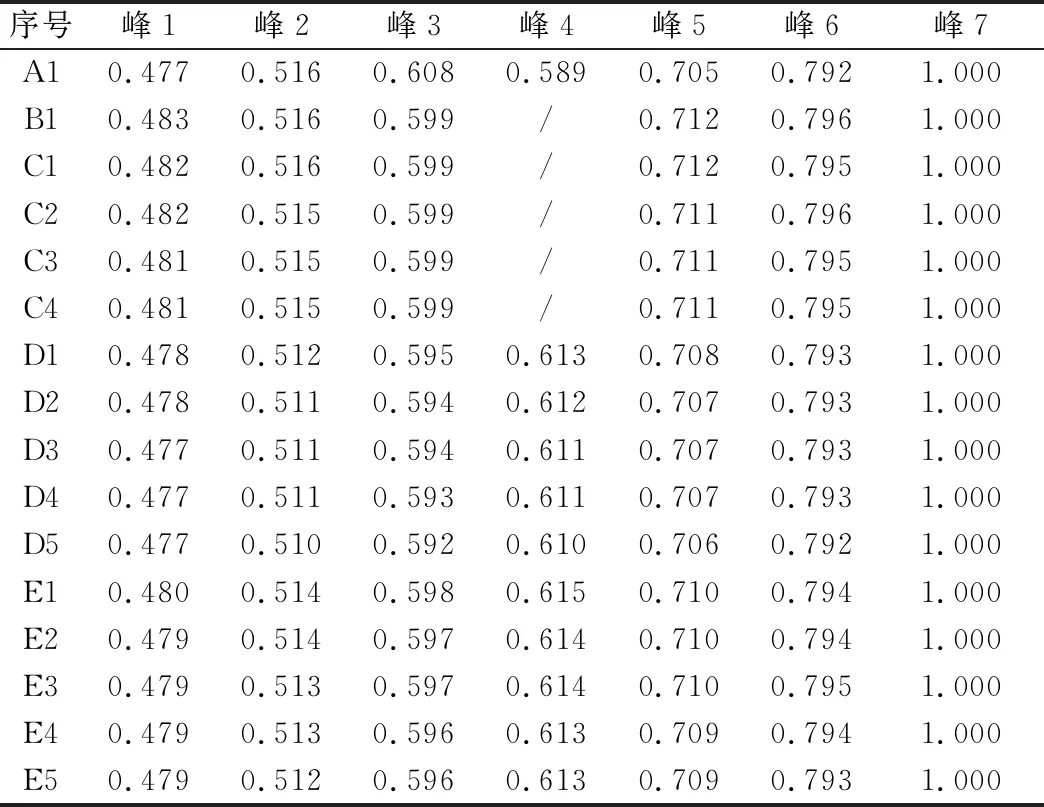
表8 百合样品特征图谱共有峰相对保留时间

表9 百合样品特征图谱共有峰相对峰面积
4 讨论
在百合药材UPLC特征图谱测定方法建立过程中,比较了不同提取溶剂(甲醇、50%甲醇、80%甲醇、乙醇、稀乙醇、80%乙醇),不同提取方法(超声、回流),不同提取时间(15、30、45 min)及不同溶剂体积(10、25、50、100 mL)对百合药材特征图谱的影响,结果表明,提取溶剂对百合药材特征峰的影响较大,以稀乙醇25 mL,超声处理(功率300 W,频率45 kHz)30 min的提取效果最佳。相比传统的HPLC,UPLC具有分析时间短、流动相消耗少、灵敏度高及分离度好等优势,为中药复杂成分的分离和解析提供了较好方法,本研究建立的UPLC特征图谱测定方法可在20 min内完成检测,操作简便,可提供较为丰富的组分群信息,综合评价百合药材的质量。
2015年版《中华人民共和国药典:一部》百合项下规定百合药材表面黄白色至淡棕黄色,有的微带紫色,断面较平坦,呈角质样。不同初加工方法百合样品的外观性状方面,直接烘干与冷水浸泡样品均为棕褐色,烫制及蒸制样品均为黄白色,但沸水烫12、16 min及蒸16 min的样品明显糊化,且干燥后呈粉性而非角质样。因此,直接烘干法及冷水浸泡法加工的样品性状不符合《中华人民共和国药典》要求,烫制2~8 min及蒸制2~12 min的样品性状符合《中华人民共和国药典》要求。文献[15]表明,百合含有大量黄酮及酚类化合物,采收后极易变色,利用高温制熟可抑制百合内活性酶的作用,防止酶促褐变,可更好保持其色泽及营养成分。因此,直接烘干及冷水浸泡的样品外观会呈棕褐色可能是未经高温制熟所致。
在不同初加工方法制备的百合样品的内在质量方面,浸出物测定结果表明,4种初加工方法百合样品的水溶性浸出物为27.09%~33.98%,醇溶性浸出物为2.59%~4.57%,无明显变化规律和差异。王百合苷B含量测定结果表明,鲜百合的王百合苷B含量为0.083 9%,直接烘干样品的含量为0.007 9%,下降显著。冷水浸泡样品的王百合苷B含量随浸泡时间增加呈明显下降趋势,当浸泡时间达6 h时,与鲜百合相比,其含量下降幅度达84.9%。烫制与蒸制样品的含量较鲜品含量均在2 min时显著升高,并在4 min后趋于平衡。烫制和蒸制样品的王百合苷B含量显著高于百合鲜品、直接烘干与冷水浸泡样品的王百合苷B含量,这可能与烫制、蒸制过程中均有高温受热有关。特征图谱测定结果表明,直接烘干及冷水浸泡百合样品均出现4号特征峰丢失,而百合鲜品、烫制和蒸制百合样品的特征图谱均正常,与不同产地18批百合药材的特征图谱基本一致,说明直接烘干法和冷水浸泡法会导致百合药材中部分特征成分丢失。因此,为保证百合药材质量,其初加工应避免使用直接烘干法及冷水浸泡法,建议选择沸水烫制4~8 min或蒸制4~12 min。
