固相萃取-超高效液相色谱法同时测定猪肉中22 种磺胺类药物残留
2021-07-28王宏宇武云龙赵栋皓孙晓亮李木子曹旭敏宋翠平赵思俊
王宏宇,武云龙,赵栋皓,孙晓亮,李木子,曹旭敏,宋翠平,赵思俊,李 存
(1.天津农学院动物科学与动物医学院,天津 300384;2.中国动物卫生与流行病学中心,山东青岛 266032;3.山西农业大学动物医学院,山西晋中 030801)
磺胺类药物(sulfonamides,SAs)是一类具有对氨基苯磺酰胺结构的药物总称,具有抗菌谱广、高效、低毒、价格低廉等特点,在兽医临床应用十分广泛。药物滥用、误用及不遵守休药期规定,会造成动物源食品中药物残留超标,既危害人类健康,还会诱导细菌产生耐药性。我国和欧盟均已制订了动物产品中的多种SAs 最高残留限量(MRL),其中我国规定SAs 总量不得超过0.1 mg/kg,奶中磺胺二甲基嘧啶不得超过0.025 mg/kg[1]。因此,对SAs 残留进行准确快速检测有着重要的食品安全意义[2]。
随着药物残留检测技术的不断发展,SAs 的前处理和检测方法越来越多。其中:前处理方法除了传统的液液萃取外,近年来还出现了许多新方法,如固相萃取(SPE)法、超声辅助分散液液微萃取法、磁性多壁碳纳米管法、基质固相分散法、QuEChERS 法等[3];检测方法主要有高效液相色谱二极管阵列检测法(HPLC-PDA)、液相色谱串联质谱法(HPLC-MS/MS)、酶联免疫法等[2,4]。目前,我国已建立了多个检测标准用于猪肉中SAs 残留检测。《畜禽肉中十六种磺胺类药物残留量的测定液相色谱-串联质谱法》(GB/T 20759—2006)中标定了牛肉、羊肉、猪肉、鸡肉和兔肉中16 种SAs 残留量的液相色谱-串联质谱测定方法,《水产品中17 种磺胺类及15 种喹诺酮类药物残留量的测定 液相色谱-串联质谱法》(农业部1077 号公告-1-2008)规定了17 种SAs 残留量的液相色谱-串联质谱法。但这些方法一次检测的药物种类较少,成本相对较高,且基质效应的存在会影响检测结果的准确度和灵敏度[5]。本研究拟采用固相萃取-超高效液相色谱法(SPE-UPLC-PDA),建立猪肉中22 种SAs 的一次性残留检测方法。
1 材料与方法
1.1 材料
1.1.1 主要仪器设备 AcquityTMUPLC-PDA 超高效液相色谱仪-二极管阵列检测器,美国Waters公司;氮吹仪,美国Organomation 公司;漩涡混合仪,美国Talboys 公司;Milli-Q 纯水仪,德国Millipore 公司;5804R 高速冷冻离心机,德国Eppendorf 公司;固相萃取装置、MCX 固相萃取柱(60 mg、3 mL),德国CNW 公司。
1.1.2 主要药品试剂 磺胺甲恶唑(sulfamethoxazole,纯 度>94%)、磺胺胍(sulfaguanidine,纯度>98%)、磺胺醋酰(sulfacetamide,纯度>97%)、磺胺吡啶(sulfapyridine,纯度>98%)、磺胺嘧啶(sulfadiazine,纯度>99%)、磺胺二甲异嘧啶(sulfisomidine,纯度>98%)、磺胺二甲恶唑(sulfamoxole,纯度>98.5%)、磺胺二甲异恶唑(sulfisoxazole,纯度>98%)、磺胺曲沙唑(sulfatroxazole,纯度>99%)、磺胺对甲氧嘧啶(sulfameter,纯度>95%)、磺胺间甲氧嘧啶(sulfamonomethoxine,纯度>96%)、磺胺甲氧吡嗪(sulfalen,纯度>97%)磺胺间二甲氧嘧啶(sulfamethazine,纯度>99%)、磺胺苯(sulfabenz,纯度>96%)、磺胺苯吡唑(sulfaphenazole,纯度>98%)、磺胺苯酰(sulfabenzamide,纯度>97%),均购自德国Dr.Ehrenstorfer GmbH 公司。磺胺(sulfonamide,纯度>99%)、磺胺喹噁啉(sulfaquinoxaline,纯度>99%)、磺胺邻二甲氧嘧啶(sulfadoxine,纯度>98%)、磺胺氯哒嗪(sulfachloropyridazine,纯度>97%),均购自Toronto Research Chemicals INC(加拿大)。磺胺二甲嘧啶(sulfamethazine,纯度>98%)、磺胺噻唑(sulfathiazole,纯度>99%)、磺胺氯吡嗪(sulfachloropyrazine,纯度>98%),均购自WITEGA Laboratorien BerlinAdlershof GmbH(加拿大)。甲醇、乙腈均为色谱纯,德国Merck 公司;甲酸、甲酸铵均为色谱纯,德国CNW 公司;乙酸、氨水均为分析纯,国药集团化学试剂有限公司;试验用水为超纯水,由Milli-Q 纯水仪制得。
1.2 方法
1.2.1 标准溶液配置 分别准确称取22 种SAs标准品于10 mL 容量瓶中,用甲醇溶解并定容至刻度,配制质量浓度为100 mg/L 的标准储备液,于-20 ℃避光保存;分别移取适量标准储备液,配制1 mg/L 的22 种SAs 混合标准储备液;分别移取适量的混合标准储备液,根据需要用甲醇逐级稀释为适当浓度的混合标准工作液,现用现配。
1.2.2 样品前处理 准确量取2 g 猪肉于50 mL具塞离心管中,加入8 mL 3%乙酸乙腈以及5 g 均质子,涡旋混匀,8 000 r/min 离心7 min,将上清移至新离心管,重复提取1 次,合并2 次上清,待净化。上清液通过经3 mL 甲醇、3 mL 水以及3 mL 3%乙酸乙腈活化的MCX 柱,分别用3 mL 水和3 mL 甲醇淋洗,用真空泵抽干后,以3 mL 5%氨化甲醇洗脱,45 ℃水浴条件下N2吹至近干,用1 mL 0.1%甲酸-乙腈(9:1,V/V)溶液复溶,涡动30 s 后,经0.22 μm 针孔式滤膜过滤,UPLCPDA 检测。
1.2.3 色谱条件 色谱柱:UPLC CSH Fluorophenyl(1.7 μm,2.1 mm×100 mm);流动相A(简称A):含有10 mmol/L 甲酸铵的0.1%甲酸水;流动相B(简称B):乙腈。流速0.3 mL/min,柱温35 ℃,二极管阵列检测器波长270 nm,进样量10 μL。梯度洗脱程序:0~2.0 min,13%B;>2.0~3.5 min,13%~15%B;>3.5~4.5 min,15%B;>4.5~6.5 min,15%~35%B;>6.5~8.8 min,35%B;>8.8~9.0 min,35%~95%B;>9.0~10.0 min,95%B;>10.0~10.2 min,95%~13%B;>10.2~12.0 min,13%B。
1.3 方法评价
1.3.1 试剂标准曲线绘制 配制质量浓度分别为2.5、5.0、10.0、20.0、50.0、100.0、200.0、250.0 μg/L的试剂标准溶液。经UPLC-PDA 进样分析,以质量浓度(μg/L)为横坐标(X),峰面积(Y)为纵坐标,绘制试剂标准曲线。
1.3.2 检测限和定量限测定 准确量取2 g 空白样品,向其中添加不同质量浓度的混合标准工作液,按照1.2.2前处理方法对样品进行处理后上机检测。以10 倍信噪比(S/N=10)对应的样品浓度作为方法的定量限(limit of quantification,LOQ),以3倍信噪比(S/N=3)对应的样品浓度作为方法的检测限(limit of detection,LOD)。
1.3.3 添加回收率和精密度测定 准确量取2 g空白样品,进行3 个水平(1、5、10 倍LOQ)的加标回收试验;按照1.2.2 前处理方法对样品进行处理后上机检测。每个水平做6 个平行,进行3 次批间重复;使用标准曲线进行校正,测定目标药物浓度,计算回收率、批内相对标准偏差和批间相对标准偏差。
1.3.4 实际样品检测 采用本试验建立的方法,对从山东省采集的18 份猪肉样品进行检测。18份样本经过3%乙酸乙腈提取、MCX 柱净化、UPLC-PDA 检测。
2 结果与讨论
2.1 色谱条件选择
由于同分异构体药物分离受限,故在流动相中加入一定量甲酸铵,使其能全部分离所有药物[6]。
磺胺类药物属于两性化合物,因此采用酸性流动相可使分离效果最佳。试验采用甲酸铵-甲酸水溶液-乙腈体系作为流动相,用0.1%甲酸-乙腈作为流动相。虽色谱峰尖锐、对称,但不能分离所有药物,故在甲酸溶液中加入适量甲酸铵进行考察。结果显示,使用含有10 mmol/L 甲酸铵的0.1%甲酸水-乙腈作为流动相,能分离出所有药物且峰形较好。为了将本试验中的药物在尽可能短的时间内有效分离,本研究采用UPLC CSH Fluoro-phenyl 色谱柱分离所有药物,采用PDA 波长270 nm,可使SAs 检测获得较高的灵敏度[7]。SAs 标准溶液色谱图、空白样品色谱图和实际样品加标色谱图分别见图1-A、图1-B、图1-C。
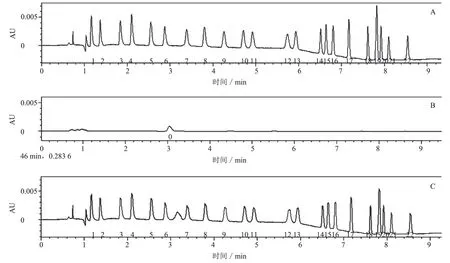
图1 标准溶液、空白样品和实际样品加标色谱图
2.2 提取液优化
提取动物产品中的SAs 通常采用乙腈、乙酸乙酯等有机溶剂或磷酸盐水溶液进行[8-9]。为了获得满意的回收率和较好的选择性,本试验选择有机溶剂和水溶液作为提取液进行优化比较。
2.2.1 有机溶剂 样品基质中的蛋白质等大分子杂质会因共萃取效应而被提取出来。由于蛋白质的最佳吸收波长为280 nm,与SAs 的检测波长较为相近,因此在紫外检测时容易影响SAs 检测的灵敏度和选择性。相关研究表明:乙腈可以沉淀牛奶等样品中的蛋白质,使蛋白质达到等电点沉淀[10];而采用乙酸乙酯和甲醇进行提取,对组织样本中目标化合物回收具有良好效果[11]。为此,本研究分别采用乙酸乙酯、甲醇和乙腈提取样品,经MCX柱净化后,使用UPLC 检测,统计目标药物回收率。结果显示:乙酸乙酯和甲醇不能完全从样品中提取药物,受基质中的内源物质干扰较大,分离不佳,且由于溶剂性质原因[12],大量脂溶性杂质溶于提取液中;而使用乙腈作为提取液时,除磺胺、磺胺醋酰外,回收率均高于80%。此外,还对乙腈的酸度进行优化分析,通过向提取液中加入乙酸或氨水等调节溶液pH。以乙腈、氨化(1%、3%、5%)乙腈、乙酸(1%、3%、5%)乙腈分别提取样本,比较不同提取液的提取效果。结果显示:使用3%乙酸乙腈,可有效提取猪肉组织中的SAs,可获得较好的回收率;增加乙酸使用量(5%),虽不影响药物回收率,但会增加萃取中杂质的干扰;而减少乙酸使用量(0、1%)时,可使SAs 回收率降低(<80%)。
2.2.2 水溶液 本试验对PBS 溶液的提取效果进行了优化,分别对PBS 溶液的pH、浓度进行优化分析。首先优化PBS 提取液pH,分别使用pH3~6的PBS 溶液进行提取。结果显示:使用PBS 溶液(pH5)时,可提取猪肉组织中的SAs,并获得了较好的回收率。增加溶液pH(pH6)时,磺胺醋酰、磺胺邻二甲氧嘧啶、磺胺间二甲氧嘧啶、磺胺二甲恶唑回收率降低(<80%);而降低溶液pH(pH3、pH4),虽不影响药物回收率,但会增加萃取中杂质的干扰,故选择pH5 的PBS 溶液。此外,还对PBS 溶液浓度进行优化分析,分别使用0.1、0.3、0.5、0.7、1.0 mol/L 的PBS 溶液(pH5)提取样本。结果显示:使用0.5 mol/L PBS(pH5)溶液,可提取猪肉组织中的SAs,并获得了较好回收率;当PBS 溶液浓度过低时,磺胺、磺胺邻二甲氧嘧啶、磺胺苯、磺胺对甲氧嘧啶的回收率小于80%;而提高PBS 溶液浓度,对目标药物回收率没有明显影响。故选择0.5 mol/L PBS(pH5)溶液作为最佳水溶液提取液。
2.2.3 两种提取液对比 对最优的有机溶剂提取液(3%乙酸乙腈)以及水溶液提取液(0.5 mol/L PBS,pH5)进行比较。分别使用两种提取液提取样本,经MCX 柱净化后,使用UPLC-PDA 检测。结果显示:以PBS 溶液、酸化乙腈作为提取液时,目标药物回收率均较高。但在试验过程中发现,以PBS 溶液作为提取液,在提取完成的待上样液中,含有大量白色絮状物,会堵塞SPE 柱,从而引起流速不稳甚至出现流不动的现象,导致试验结果平行性较差[13]。因此,本研究选择3%乙酸乙腈作为猪肉样品提取液。
2.3 净化条件优化
2.3.1 SPE 固相萃取柱选择 本研究选取1#(CNW-HLB 柱)、2#(Oasis HLB 柱)、3#(CNW-MCX 柱)、4#(PCX 柱)进行比较。标准添加量为50 μg/kg,经甲醇、水和上样液活化的SPE 柱富集,用甲醇或5%氨化甲醇洗脱,比较各SPE 柱对目标药物的富集效果。结果显示,1~4#的富集比例分别为90%、85%、90%和83%。结果表明,4 种SPE 柱均可富集SAs,且CNW 公司生产的HLB 柱(1#)以及MCX 柱(3#),对目标药物的富集效果较好。考虑到在试验过程中,由于提取液为乙腈,只有在过HLB 柱前氮吹至近干,再使用复溶液复溶后,才可继续进行净化步骤,因而容易导致目标药物损失[14],且会浪费大量时间;而MCX 柱,由于其为阳离子交换柱,对pH 较为敏感,以3%乙酸乙腈作为提取液时,可以直接上样。故选择3#作为SPE 固相萃取柱。
2.3.2 洗脱液优化 按参考文献[15-16],选择4种洗脱液,1%氨化甲醇、3%氨化甲醇、5%氨化甲醇、7%氨化甲醇,各取3 mL 洗脱液过已富集的MCX 固相萃取柱,比较洗脱效果。结果显示:5%和7%的氨化甲醇洗脱能力无明显差异(>95%),可完全将药物从柱床中洗脱下来,1%氨化甲醇仅可洗脱下20%~40%,而3%氨化甲醇可洗脱下60%~80%,故选择5%氨化甲醇作为洗脱溶液。
2.4 方法学考察
2.4.1 线性范围、相关系数、检出限和定量限以不同浓度的混合标准溶液进行测定,以质量浓度为横坐标(X),峰面积为纵坐标(Y),绘制试剂标准曲线。结果(表1)显示:各药物线性关系良好,相关系数R2>0.999。LOQ 为2.5~10.0 μg/kg,LOD为1.0~4.0 μg/kg。
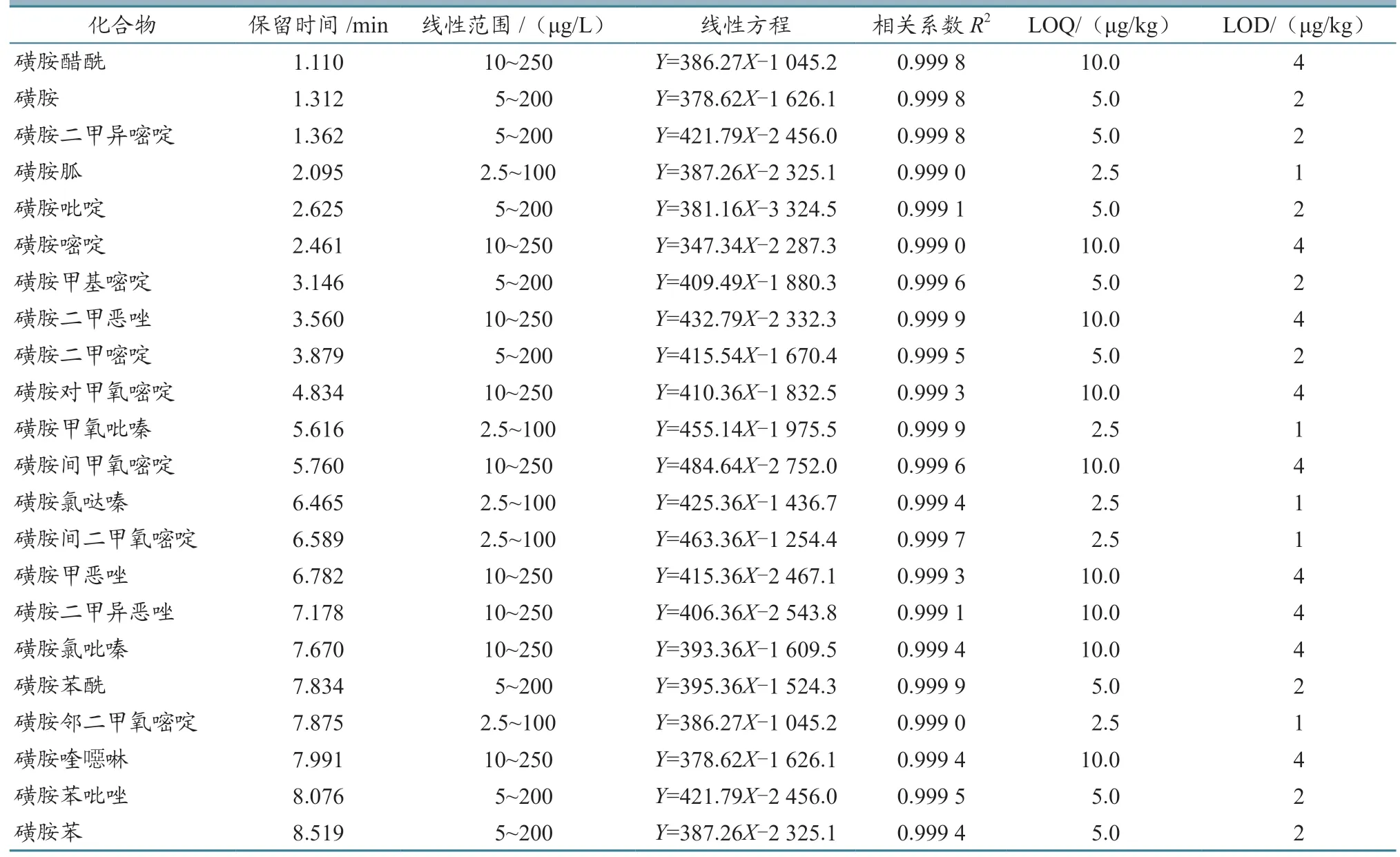
表1 22 种兽药的保留时间、线性方程、相关系数和LOQ、LOD
2.4.2 准确度和精密度 22 种目标药物回收率及批内、批间相对标准偏差见表2。结果显示,回收率为80.4%~96.6%,批内相对标准偏差为1.1%~4.6%,批间相对标准偏差为2.5%~13.4%,可满足国际上对兽药残留的检测需求。

表2 22 种兽药的加标回收率和相对标准偏差
2.5 相对于UPLC-MS/MS 的优势
在使用UPLC-MS/MS 法时,基质效应(matrix effects,ME)的存在会影响检测结果的准确度和灵敏度[17]。经前处理样品中加入目标药物,进行UPLC-MS/MS 检测,绘制基质匹配标准曲线,通过计算各药物样本溶液与浓度相同的标准溶液之比的平均值,计算目标药物的基质效应[18]。结果显示:目标药物中有部分基质效应较强(ME <60%),包括磺胺对甲氧嘧啶、磺胺二甲基异嘧啶、磺胺甲基嘧啶、磺胺二甲基嘧啶;部分基质效应很强(ME <40%),包括磺胺邻二甲氧嘧啶、磺胺间甲氧嘧啶、磺胺苯。基质效应严重影响结果的准确度和灵敏度,且UPLC-MS/MS 成本较高,操作复杂。而本研究的UPLC-PDA 法不存在基质效应影响,在经前处理后杂质较少,且成本较低、操作简便,结果准确性较高,重复性良好。
2.6 实际样品检测
利用本研究建立的方法对从山东省采集的18份猪肉样品进行测定,结果未有SAs 检出。
3 小结
本研究建立的猪肉中22 种SAs 的SPE-UPLCPDA 检测方法灵敏度高、重复性好、成本低廉,且操作简单、快速,可以用于检测猪肉样品中的SAs 残留检测。
