新金色分枝杆菌CRISPR 基因编辑技术的构建及其初步应用
2021-07-28童丽珍杨套伟邵明龙徐美娟廖祥儒饶志明
林 春 , 童丽珍 , 杨套伟 , 邵明龙 ,张 显 , 徐美娟 , 廖祥儒 *, 饶志明 ,
(1. 江南大学 工业生物技术教育部重点实验室, 江苏 无锡 214122;2. 江南大学 生物工程学院, 江苏 无锡214122)
关键字: 新金色分枝杆菌;CRISPR;基因编辑;转录激活
近年来, 新金色分枝杆菌(Mycobacterium neoaurum) 已成为甾体激素类药物的生物合成平台之一。 甾体药物被广泛应用于治疗风湿、心血管疾病、淋巴性白血病、人体器官移植、抗肿瘤、细菌性脑炎、皮肤病、内分泌失调等疾病[1]。据统计,2011 年甾体激素药物销售额已高于280 亿美元,约占全球医药总销售额的6%,成为产量第二大类药物,仅次于抗生素[2-3]。 随着甾体药物价格的稳定,采用生物技术生产甾体药物逐渐成为主流。 作者所在实验室前期筛选得到了一株甾体药物的高产菌株新金色分枝杆菌, 并对该菌株的研究取得了一定的进展,然而进一步调控新金色分枝杆菌甾醇代谢方式急需探索出新的基因编辑技术。
目前应用于分枝杆菌基因编辑的技术主要依赖于等位基因交换、 同源重组和非黏性末端修复。等位基因交换是两步重组过程:第一步,使用抗生素抗性标记分离携带整合到宿主基因组中的整个递送载体的单交换;第二步,分离双交换,其可以具有两个基因等位基因中的任一个,以去除标记[4]。 通过同源重组获得目的基因敲除突变株也是对分枝杆菌进行基因编辑的有力工具。 但是,分枝杆菌同源重组的发生率比其他细菌明显低很多,仅为10-6~10-5。利用广泛的自杀式载体pNIL/pGOAL 系列质粒用于构建无标记缺失突变菌株也可以实现目的基因的阻断或者敲除[5]。NHEJ 是 DNA 双链断裂(DSB)的主要修复途径,它往往容易在连接位点插入或缺失突变,并产生破坏靶基因的移码突变。 与同源重组相比,NHEJ 系统不依赖于重组蛋白质或同源DNA 模板,它是高等生物维持基因组稳定性的主要DNA 修复系统[6]。Gupta 等报道了 NHEJ 介导的染色体I-Sce I 诱导的DSB 的修复,修复效率为25%。然而,双I-Sce I 位点首先必须置于靶基因内,并且ISce I 引入编码质粒以产DSB,总基因组编辑周期约为15 d[7]。
近年来随着基因编辑的新型魔术刀——CRISPR (Clustered regularly interspaced short palindromic repeats)编辑技术的问世,为微生物的基因工程改造提供了新手段,CRISPR/Cas 系统的主要功能元件有两部分:其一是用于切割目标靶点DNA的Cas 核酸酶; 另一个是能与标靶DNA 互补配对的 小 型 向 导 RNA (sgRNA)[9]。 其 中 基 于 Ⅱ 型CRISPR/Cas 系统研发作为切割DNA 双链的工具已经获得研究者们的青睐,并且已经成功应用于大肠杆菌、斑马鱼、拟南芥、酿酒酵母等物种的基因编辑中[8]。 Cas 是一种 DNA 核酸酶,位于 CRISPR 序列旁边,RNA 引导的核酸酶可以识别和切割靶基因以产生DNA 断裂。 在没有DNA 模板的情况下,可以通过在相应的同源模板或NHEJ 存在下的同源重组来修复断裂[10]。 最近,研究者已经证明CRISPR/dCas9(失活的Cas 蛋白)可以控制耻垢分枝杆菌和结核分枝杆菌中的基因表达[11-12]。 有研究表明,CRISPRCas12a 辅助的同源重组系统进行耻垢分枝杆菌的基因组编辑时, 引入位点直接突变的效率高达80%,而结合双链模板DNA 的重组、删除或者插入效率可达到 37%~75%[11,13]。
本研究是自主构建CRISPR/Cas 系统, 通过多方式的改进以两质粒体系构建用于新金色分枝杆菌的基因编辑。 为确保质粒在分枝杆菌中能稳定复制和遗传,采用了含有分枝杆菌来源的复制子OriM的表达载体pMV261 作为骨架结构;并对3 种不同性质的Caseffactor 蛋白质进行比较分析, 使用了和新金色分枝杆菌同源性较高的谷氨酸棒杆菌基因组优化的 spCas9cg 作为 Caseffactor, 其 GC 含量为53%, 与新金色分枝杆菌基因组的GC 含量62%相近。 作者选取了Mycobacterium neoaurumJC-12 菌株甾醇代谢途径的关键基因ksdd作为实现基因组水平的基因编辑目标位点,首先通过PCR 验证以及测序验证初步测定了基因的敲除, 再通过RT-qPCR、HPLC 以及酶活检测定量评价了此编辑系统。 结果显示,经初步优化后的CRISPR/Cas9 系统可应用于新金色分枝杆菌的基因组编辑,该系统也显示出在非模式菌株中应用的潜力,为诸多野生型菌株提供了代谢工程改造的参考。
1 材料与方法
1.1 菌株与质粒
M.neoaurumJC-12: 作者所在实验室筛选并保存; 大肠杆菌JM109 和DE3 以及分枝杆菌表达载体pMV261:作者所在实验室保存;其余菌株及实验中所用的质粒:见表1。
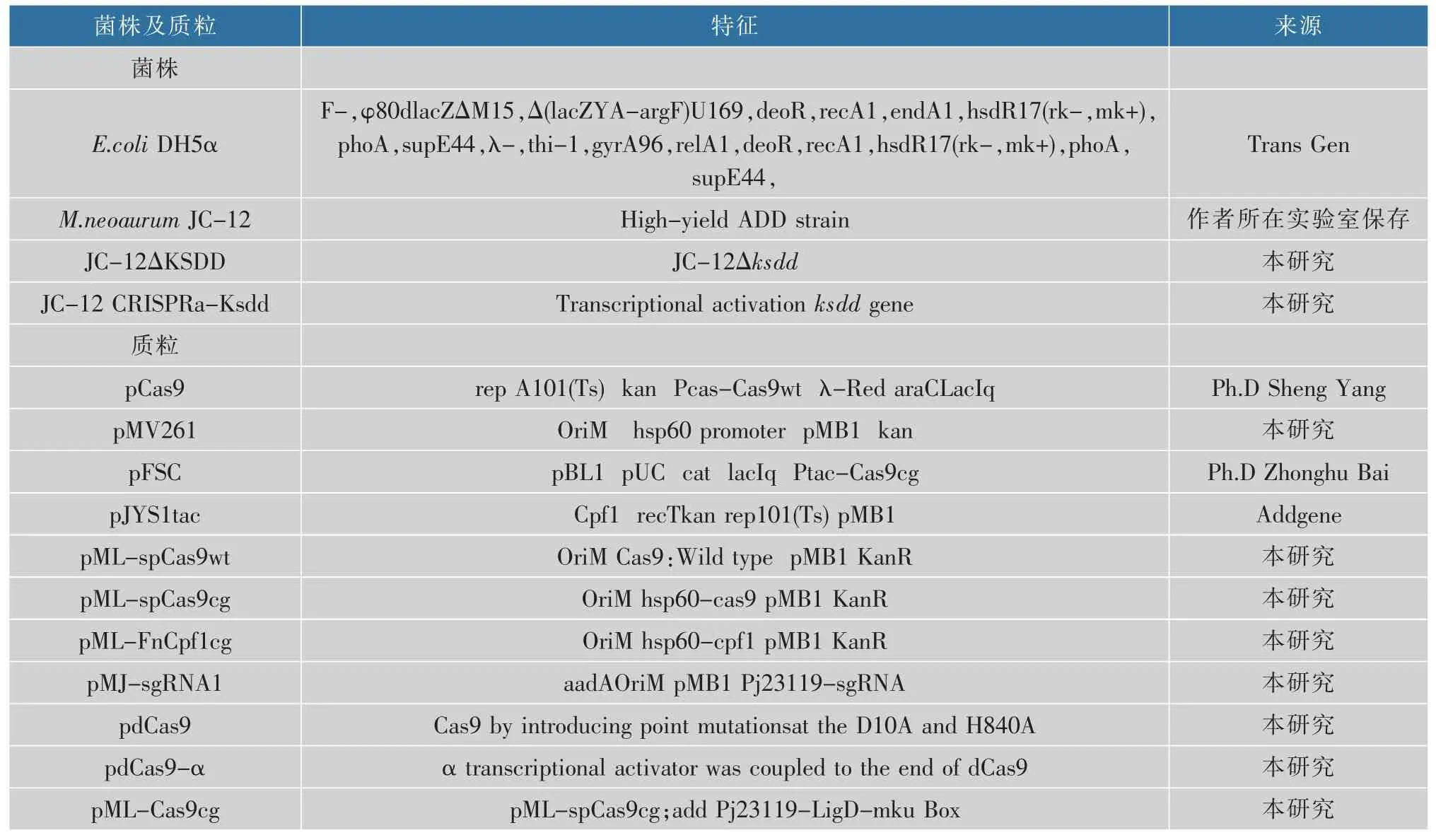
表1 本研究所用的菌株和质粒Table 1 Strains and plasmids used in this study
1.2 菌株的培养
1.2.1 大肠杆菌的培养 LB 培养基(g/L):蛋白胨10,酵母提取物 5,NaCl 10;如需固体培养基,加入质量分数2%的琼脂。 旋转式摇床180 r/min、37 ℃。转化子筛选条件:加入卡那霉素至终质量浓度为50 μg/mL,氯霉素为 10 μg/mL,链霉素为 100 μg/mL。
1.2.2 新金色分枝杆菌的培养 种子培养基(g/L):葡萄糖 20,蛋白胨 5,牛肉膏 5,甘油 15,NaCl 15,pH 7.0;摇瓶发酵培养基(g/L):葡萄糖 20,蛋白胨10,K2HPO4·3H2O 3,MnCl20.000 5,MgSO4·7H2O 0.2,植物甾醇 10,环糊精 30,pH 7.0(250 mL 三角瓶,装液量 50 mL);感受态制备培养基:种子培养基+体积分数0.2% Tween 80。 旋转式摇床转速为180 r/min、30 ℃。 转化子筛选条件:加入卡那霉素终质量浓度为 30 μg/mL,氯霉素为 10 μg/mL,链霉素为 30 μg/mL。
1.2.3 甾醇产量的测定 新金色分枝杆菌JC-12降解植物甾醇的主要产物是 AD 和 ADD,AD 和ADD 在254 nm 处均有吸收峰, 因此可采用高效液相色谱(HPLC)检测,具体测定方式参照文献[14]。
1.2.4 KSDD 酶活的测定 采用PMS-DCPIP 法测定 KSDD 酶活[14]。 KSDD 脱去底物 AD 的两个氢生成ADD, 蛋白质自身的FAD 接受脱去的 2H+和2e-成 FADH2,然后 FADH2 将 DCPIP 还原,DCPIP 在600 nm 处有吸收峰。3 mL 反应混合物由 100 μL 粗酶液、50 mmol/L 的 Tris-HCl(pH 7.0)、40 μmol/L 的DCPIP、1.5 mmol/L 的 PMS、500 μmol/L AD(溶于体积分数2%的甲醇)所组成,检测600 nm 处的吸光值变化。 酶活单位定义:将在1 min 内还原1 μmol DCPIP 所需的酶量定义为一个酶活单位(U)。
1.3 主要试剂与仪器
基因组提取试剂盒、质粒提取试剂盒、胶回收试剂盒:购自上海捷瑞生物工程有限公司;抗生素、阿拉伯糖、2,6-二氯酚靛酚(DCPIP)及吩嗪硫酸甲酯(PMS)等:购自阿拉丁试剂公司;Taq 酶、限制性内切酶、T4 DNA 连接酶等:购自TaKaRa 公司;植物甾醇(大豆甾醇质量分数≥95%):购自礼来生物技术 (湖州) 有限公司;ADD 及 AD 标准样品: 购自Sigma 公司; 一步法克隆试剂盒、RT-qPCR 试剂盒及相关用品: 购自南京诺唯赞生物科技有限公司;PCR 仪:购自 Eppendorf Mastercycler nexus X2;电转化仪:购自 BIO-RAD pulse controller(Gene Pulser);荧光定量 PCR 仪: 购自 StepOnePlus;HPLC 高效液相色谱仪:购自岛津LC-20AB。
1.4 质粒的构建与基因的克隆
1.4.1 CasEffactor 的选择与表达载体的构建 以pCas9 为模板,P1/P2 为引物克隆出 wtCas9; 以 pC为模板,P3/P4 为引物克隆出Sp-Cas9cg (为谷氨酸棒杆菌密码子优化的Cas9 基因); 以pJYS1 为模板,P5/P6 为引物克隆出Fn-Cpf1cg 基因;以上基因的克隆均用高保真酶Phanta Max Master Mix (购自南京 Vazyme), 再将BamH I 线性化 pMV261 载体与 Cas Factor 表达框架通过 ClonExpress II One Step Cloning Kit(购自南京Vazyme)构建组成型的表达Cas 效应蛋白质的载体 pMV261-spCas9wt、pMV261-spCas9cg、pMV261-FnCpf1。 通过苏州金唯智生物科技有限公司合成了LigD和mku基因。 通过P7/P8 和P9/P10 引物将LigD和mku基因融合成Pj23119-LigD-mku-rrnB 表达框架, 再通过 Clon-Express II One Step Cloning Kit 将 NHEJ 表达框架构建到Cas 表达载体上得到pML-Cas9。
1.4.2 sgRNA 载体的构建 以ptrc99A-Spc 载体为基本骨架, 以P11/P12 从pMV261 载体上克隆得到OriM, 以 P13/P14 克隆得到 sgRNA 框架 168 bp,ClonExpressMultiS One Step Cloning Kit 将 OriM、sgRNA 连接得到 pMJ-sgRNA。 通过根据靶序列sgRNA 软件 CRISPR/Cas9 gRNA Finder 设计靶向ksdd基因CDS 区的sgRNA 以及靶向启动子区域的sgRNA。通过P15/P16 引物以pMJ-sgRNA 为模板克隆出靶向ksdd编码区的pMJ-sgRNA,命名为pMJ-sgRNA1-ksdd,通过 P17/P18 引物 pMJ-sgRNA 为模板克隆出靶向ksdd启动子区的pMJ-sgRNA, 命名为pMJ-sgRNA2-ksdd。 将纯化后的PCR 产物通过Solution I(Takara,上海)连接后转化到E.coliDH5α中,并通过测序鉴定sgRNA 的N20 序列,所用引物见表2。
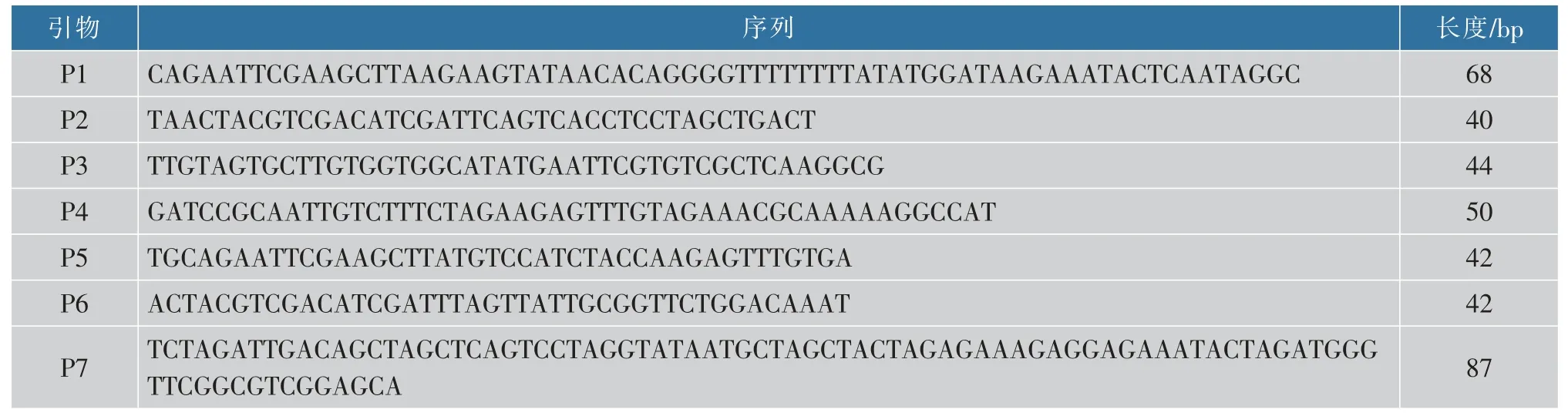
表2 本研究中所用的引物Table 2 Primers used in this study

续表2
1.5 运用CRISPR 技术介导的基因编辑
1.5.1 CRISPR/Cas 介导的分枝杆菌基因组基因的敲除 新金色分枝杆菌JC-12 用于构建突变菌株,其感受态的制作方法以及电转方法见文献[14]。 首先将构建完整的pMLcas9 载体转化到新金色分枝杆菌JC-12 中, 通过PCR 验证挑取含有 pMLcas9的转化子,将其转接到含有3 μg/mL 卡那霉素的分枝杆菌感受态种子培养基中,30 ℃、180 r/min 培养48 h, 再转接到分枝杆菌感受态培养基中培养6~8 h 至OD 为0.6~0.8,回收菌体制备电转感受态。电转时将300 ng 的pMJ-sgRNA1 载体加到感受态细胞中,轻柔混匀,冰上放置30 min 后加到预冷的1 mm电转杯中, 在2.2 kV 的条件下放入电转仪 (Bio-Rad)中电转,向电击杯中加入900 μL 液体培养基,混匀并全部转移至EP 管中,30 ℃摇床复苏4~6 h;离心收集细胞,去除部分上清液,涂布于含30 μg/mL卡那霉素和30 μg/mL 链霉素抗性的固体平板上,30 ℃培养箱倒置培养5~7 d,转化子用PCR 验证。
1.5.2 利用CRISPR-activate 调控分枝杆菌基因组水平的表达 Cas9 核酸酶剪切活性取决于结构域RuvC 和HNH。这两个结构域分别负责切割DNA 链的两条链, 通过对这两个位点定点突变, 即得到dCas9(D10A&H840A),命名为 pdCas9。 从新金色分枝杆菌中用P23/P24 克隆得到转录激活因子α 亚基,然后利用Gibson 组装通过linker(GGTGGCGGA GGGT CAGGTGGCGGA)将转录激活因子α 亚基偶联到 dCas9 末端,得到 pdCas9-α 载体。
2 结果与分析
2.1 CRISPR-Cas 系统质粒的构建
按照上述方法将扩增得到的基因片段进行融合连接转化,挑取转化子提取质粒后,将质粒进行PCR 验证,出现特异性条带,大小正确,后续的重组质粒测序结果正确,见图1。
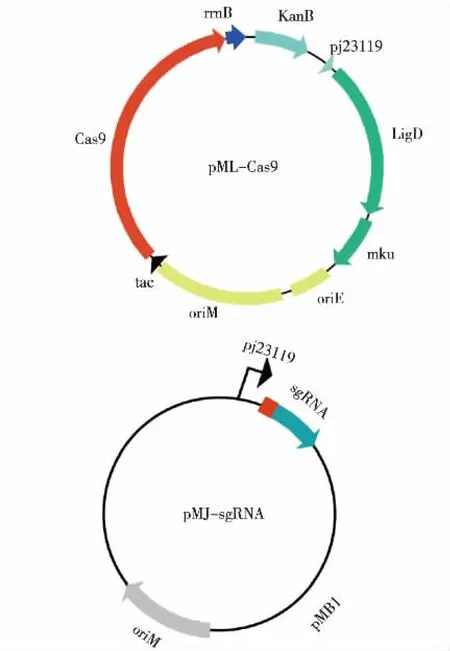
图1 新金色分枝杆菌CRISPR-Cas 双质粒基因编辑系统Fig. 1 CRISPR-Cas double plasmid system of Mycobacterium neoaurum
2.2 CRISPR-Cas 系统在新金色分枝杆菌中的表达与稳定性分析
将上述构建完整的pML-SpCas9wt、pML-SpCas9cg和pML-FnCpf1cg载体分别电转到新金色分枝杆菌JC-12 中, 通过抗性平板筛选出转化子进行菌落PCR 验证,分别对Caseffactor 表达框架用相应的引物进行验证,见图2。 电泳结果证明,上述质粒均已成功转入新金色分枝杆菌中,进一步通过RT-qPCR验证了 Caseffactor 和sgRNA 的转录。 研究发现,野生型Cas9 质粒在新金色分枝杆菌中不能稳定遗传。 为进一步探索不同Caseffactor 对新金色分枝杆菌的菌株的影响,作者分析了同等条件下对新金色分枝杆菌生长的影响,结果见图3。 发现spCas9wt对新金色分枝杆菌的生长是有影响的, 而spCas9cg和FnCpf1cg对生长的影响相对较小, 因而后期实验中采用spCas9cg作为本研究的Caseffactor。
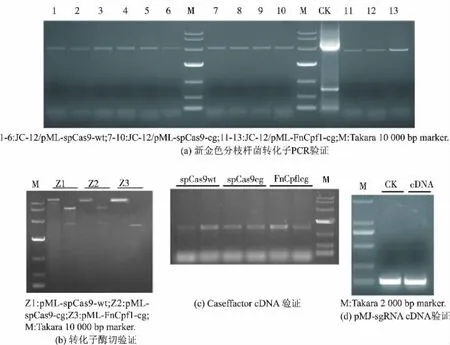
图2 新金色分枝杆菌Caseffactor 转化子验证与RT-PCR 验证Fig. 2 Caseffactor transformant validation and RT-PCR validation of Mycobacterium neoaurum
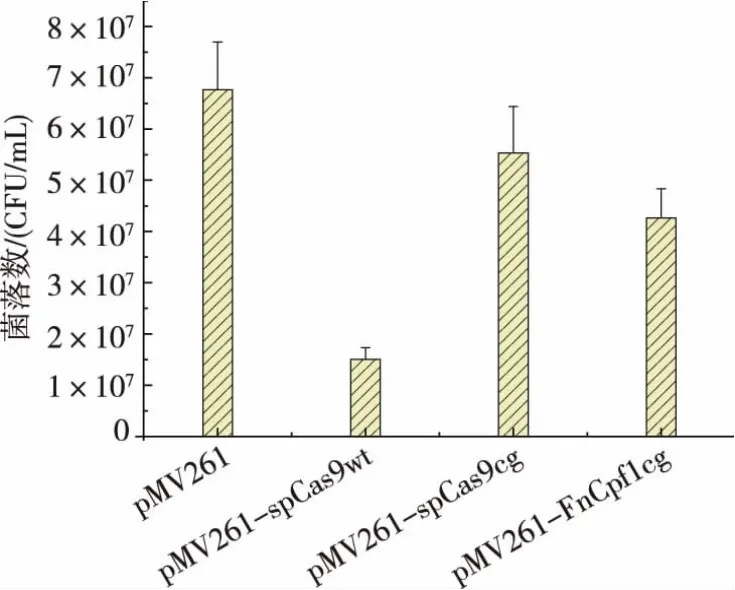
图3 不同Caseffector 对新金色分枝杆菌生长的影响Fig. 3 Effect of different caseffector on the growth of Mycobacterium neoaurum
2.3 基因突变菌株的鉴定
将上述构建好的Caseffactor 表达载体和sgRNA 载体按1.5.1 的方法转入到新金色分枝杆菌中, 筛选得到转化子后, 挑取对应的单菌落并用P27/P28 引物进行PCR 验证,初步判定敲除了ksdd基因,然后挑取单克隆于30 ℃、180 r/min 培养5 d,用细菌DNA 提取试剂盒对突变株提取基因组DNA,以突变株基因组DNA 为模板,同样用上述引物进行扩增, 获得了敲除ksdd基因成功的菌株,并将PCR 产物送至金维智生物科技有限公司测序,结果见图4。
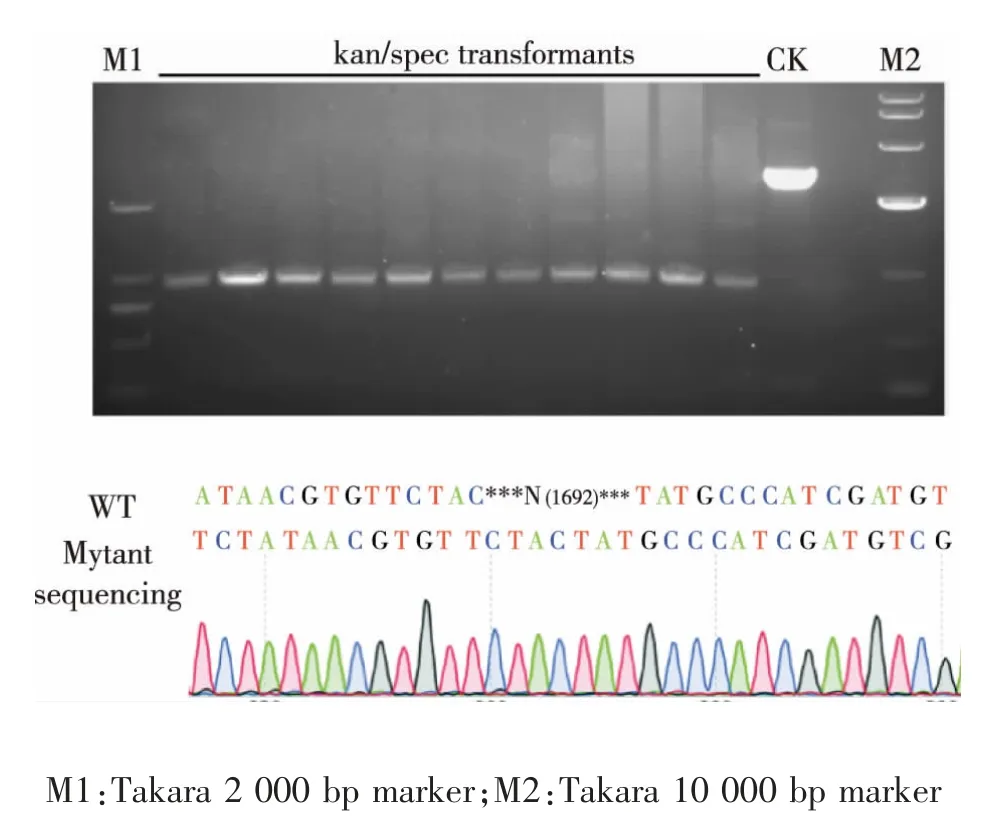
图4 突变菌株的PCR 验证图与测序结果Fig. 4 Verification and sequencing results of mutant strains
2.4 突变菌株的生长与甾醇转化效率及酶活的分析
对上述验证正确的敲除菌株转接至种子培养基中培养5 d,测定菌株的生长曲线变化。 如图5 所示,突变型和野生型新金色分枝杆菌在生长上无显著差异。 将KSDD 突变菌株转接到摇瓶发酵培养基中发酵培养 5 d, 在48 h 和96 h 取样测定ADD 和AD 的产量,结果见图6—7。ksdd敲除菌株已经完全不产ADD,而且AD 的产量相比于野生型菌株提高了25%左右。 突变株的发酵结果也间接反映了CRISPR/Cas9 系统在新金色分枝杆菌中成功应用于基因组的敲除。 同时对突变株的KSDD 的酶活进行了测定,结果见图8。 突变株Mutksdd的酶活下降了80%左右。
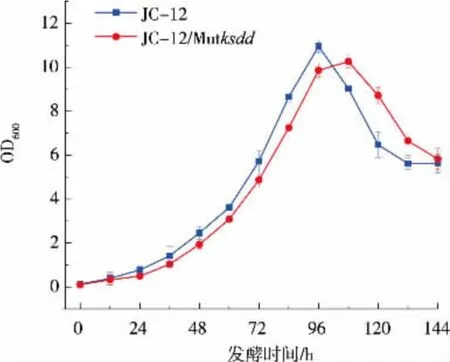
图5 突变菌株生长曲线Fig. 5 Growth curve of mutant strain
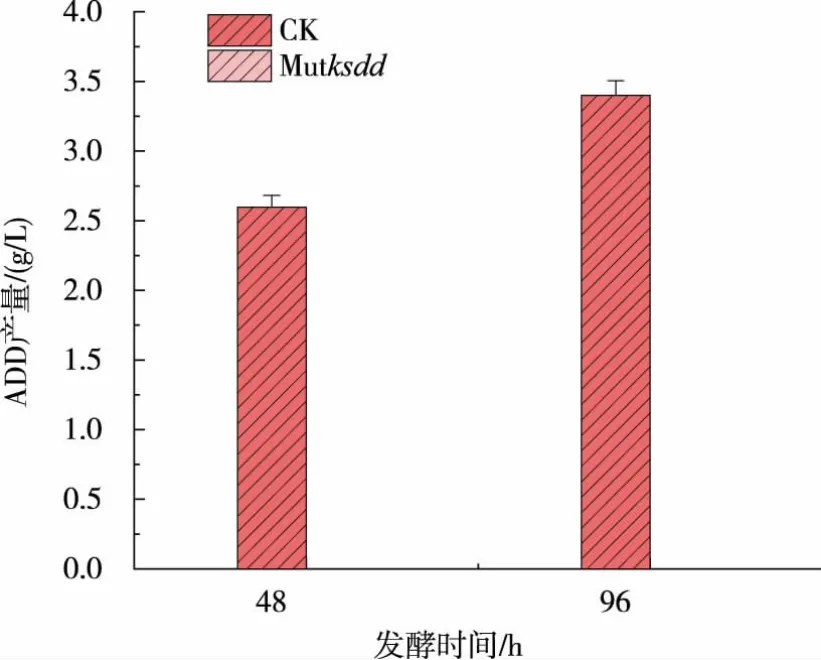
图6 ADD 产量对比Fig. 6 Comparison with the production yield of ADD
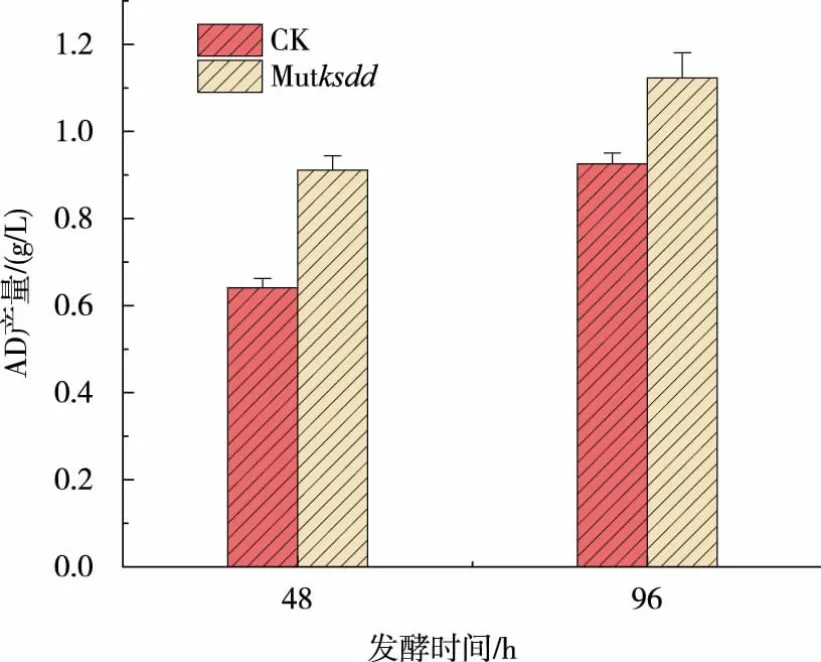
图7 AD 产量对比Fig. 7 Comparison with the production yield of AD
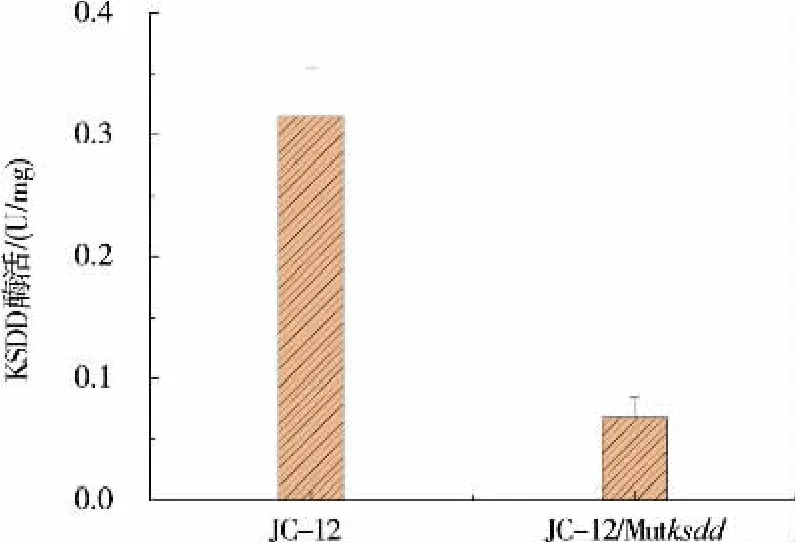
图8 KSDD 酶活的对比Fig. 8 Comparison of the KSDD activity
2.5 ksdd 在不同转录激活因子下的表达调控分析
将突变后的dCas9 与RNA 聚合酶α 亚基偶联后转入到新金色分枝杆菌中,针对ksdd起始密码子前-29、-83、-141 位置的设计 3 个 sgRNA, 分别命名为 Ca-g1、Ca-g2、Ca-g3,并将其转入到含 dCas9-α质粒的新金色分枝杆菌中。 筛选得到转化子后将转化子接种到种子培养基中培养5 d, 取样用于RT-qPCR 实验,RT-qPCR 实验结果见图 9。 dCas9-α 对ksdd转录水平的调控有显著的影响, 其中靶向于-29 位点对转录产生了一定程度的抑制,而靶向-83 位点和-141 位点则对ksdd的转录水平有了显著提升。 同时对不同调控位点对甾醇产物的变化进行了96 h 发酵分析, 对ksdd的转录调控使得AD 和ADD 的产量在-83 位点分别提升了26%和25%,在-141 位点分别提升了29.5%和36%, 而靶向-29位点却无显著变化,这与不同靶点的ksdd的表达水平也是吻合的,见图10。 但总体而言,对ksdd的调控未达到理想效果,具体原因可能需进一步优化和改进此CRISPRa 体系。
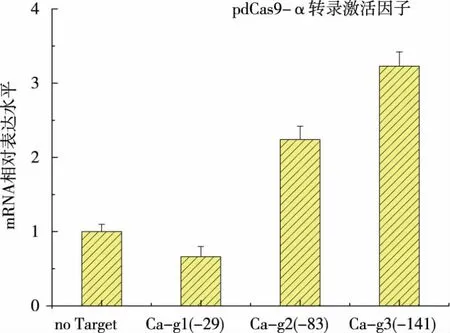
图9 CRISPRa 对ksdd 的转录水平的变化Fig. 9 Transcription changes of ksdd regulated by CRISPRa
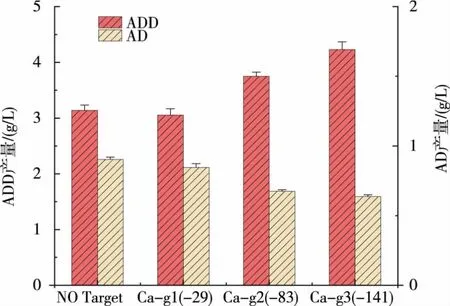
图10 CRISPRa 对甾醇产物的变化Fig. 10 Transcription changes of sterol products regulated by CRISPRa
3 结 语
作者构建了可在新金色分枝杆菌这一重要甾醇药物合成微生物基因组中实现敲除和插入等编辑功能的CRISPR/Cas9 系统,基于该系统实现了对新金色分枝杆菌甾醇药物代谢途径中的重要中间体AD 和ADD 合成的关键基因ksdd的单基因敲除与基因组水平的表达调控,该系统参考相关类似的研究[15-16],以分枝杆菌表达载体pMV261,该载体含有一个分枝杆菌复制原点OriM 和大肠杆菌复制原点OriE,能够在大肠杆菌和分枝杆菌中稳定复制和表达; 以此载体为骨架分别用Pj23119 和tac 启动子表达了NHEJ 修复框架和Cas9 表达框架,并重新构建了一个含有OriM 的在强启动子下sgRNA 表达载体。 将双质粒体系导入到新金色分枝杆菌中,表达产生的Cas9 和转录形成的目标靶向sgRNA 结合靶向目标靶点对其进行切割,致使基因组形成双链DNA 断裂(Double strand breakage,DSB),绝大多数细胞因此死亡。 由于介入了NHEJ 修复从而激活NHEJ 的修复,Ku 蛋白复合物识别结合到DSBs 末端, 再招募 DNAligase4 (LigD) 将 DSB 末端连接。NHEJ 虽能高效连接修复断裂DNA, 但是也能造成不确定性的缺失从而导致基因的缺失。 而转录激活是参考CRISPR-Cas9 的这种形式引入转录激活因子, 从而招募更多的RNA 聚合酶使得靶向基因的表达水平提高[17]。 作者构建的CRISPR-Cas9 系统初步实现了新金色分枝杆菌的基因编辑,开拓了新金色分枝杆菌的基因编辑方式,从而为进一步探索和提升分枝杆菌甾醇代谢途径和相关基因提供了依据。
虽然已经实现了对新金色分枝杆菌的基因敲除,但是未达到理想效果。 首先未对Cas9 的表达在新金色分枝杆菌中做相对的优化以及对NHEJ 的修复效率做进一步的提升,初期实验筛选过程中出现未敲除菌株和敲除菌株的混合菌落,有其他研究者也出现过类似情况[18],究其原因是大多数原核微生物缺乏DSB 断裂修复方式导致产生DNA 断裂的菌株不能修复而死亡,使得脱靶的菌株能够以优势菌存活,通过引入HDR 或者NHEJ 修复可以显著提升敲除菌株的存活;其次对于sgRNA 的脱靶率是该领域一直探索的问题,本研究中为降低脱靶效应带来的影响,参考了相关的研究[19],在设计靶向目标靶点区域的序列设计多个sgRNA。
近年来随着研究者在Cas 蛋白的功能与新型Cas 蛋白的研究中发现, 研究者Choudhary. Eira 等人通过对化脓性链球菌原始dCas9 中的191 个密码子进行修饰以及采用四环素诱导型启动子Pmyctet0, 使得dCas9 稳定表达同时降低了dCas9对菌体生长的影响[11]。 Jeremy M. Rock 等人通过对11 种不同来源的Cas9 蛋白的筛选, 鉴定出其中4种对分枝杆菌靶向基因敲除是广泛适用的,而来自嗜 热 链 球 菌 (Streptococcus thermophilus) 的CRISPR1 Cas9(dCas9Sth1)介导的基因敲除率低,是稳定的且具有最小的蛋白质毒性[13]。 2015 年张锋团队发掘了 CRISPR 家族的新成员—Cas12a(Cpf1),随后Mei-Yi Yan 等人使用CRISPR-Cas12a 辅助的同源重组系统进行耻垢分枝杆菌的基因组编辑时发现,通过四环素诱导表达的Cpf1 蛋白对耻垢分枝杆菌的生长并无显著影响[12]。 这些研究也将为进一步完善新金色分枝杆菌CRISPR/Cas 系统提供参考依据, 进一步改进和应用其他编辑效率更高的Cas蛋白,完善以及提升新金色分枝杆菌的CRISPR/Cas基因编辑工具,为新金色分枝杆菌的基因组水平的研究和功能探索提供高效的方法。
