慢羽鸡PRLR 和SPEF2 基因连接方式和融合基因双向转录研究
2021-07-24白少川王德贺葛琳涵郭艳丽樊宝良李兰会
白少川,李 楠,王德贺,葛琳涵,郭艳丽,樊宝良,李兰会
(河北农业大学 动物科技学院,河北 保定 071001)
Sevebrovsky[1]在1922 年首次提出雏鸡主翼羽与覆主翼羽生长速度不同形成的快羽和慢羽表型为伴性性状,受1 对等位基因K 和k+控制,并且慢羽对快羽为不完全显性[2]。羽型被广泛应用到鸡配套系生产的商品代性别鉴定上,且不存在显性白基因的上位效应,使之应用比羽色更广[3]。在核心育种群中需要纯合快慢羽鸡,其中母鸡的表型直接反映了基因型,而纯合公鸡ZKZK或杂合公鸡ZKZk+均表现为慢羽。无论是建立专门的品系还是血统纯化过程中,纯合慢羽公鸡的选择,都是必不可少的。近年来,国内地方品种相继建立羽型自别雌雄的配套系生产,但不同品种鸡的羽型对生产性能的效应有一定差异[4-5]。
慢羽K 基因具有复杂的分子结构[6-7],该区域不仅包含k+ 位点中分别位于Z 染色体(NC_006127.5)的催乳素受体基因(PRLR)和精子鞭毛2 基因(SPEF2),还存在由于同源重组形成的部分重复基因dPRLR和dSPEF2。张秀玲[8]利用重复基因的断裂连接点建立了慢羽公鸡羽型基因型的半定量PCR 检测方法。Iraqi 等[9]和Elferink等[6]分别通过Southern blot 和定量PCR 认为K基因有180 kb 的串联重复序列,但慢羽PRLR、SPEF2、dPRLR和dSPEF2等4 个基因的排列构成未见试验报道。PRLR和SPEF2基因5'末端以“头碰头”形式连接,并有长477 bp 的共有区域;dPRLR与dSPEF2的3'末端“尾对尾”连接形成融合基因,推断4 个基因的排列构成。融合基因由SPEF2内含子4 的1 546 bp 处与PRLR第12 外显子的578 bp 处连接形成。Zhao 等[10]发现融合基因具有双向转录特征。Ayako 等[11]发现dSPEF2基因的多个转录本,其4 个新的外显子位于dPRLR反义链的内含子区;dPRLR基因3'末端的4 个polyA 位于dSPEF2反义链的内含子区。
随着高通量测序技术的发展,越来越多的研究发现动植物基因组中很多基因同时存在正义转录本和反义转录本[12-14]。反义转录物参与早期胚胎发育过程中的细胞增殖与迁移[15],以及癌变调控[16-18]。由于反义转录物可与互补的正义链形成双链RNA,导致RNA 干扰、RNA 编辑或RNA 掩蔽[19],对邻近基因的表达产生影响[20]。
本试验通过半定量PCR 检测PRLR和SPEF2基因5'末端共有区域477 bp 片段拷贝数,明确慢羽K位点基因的连接方式;通过特异性反转录确定共有区域477 bp 片段和融合基因的转录特征,以期为深入挖掘慢羽鸡K 基因的分子结构和调控机制奠定了基础。
1 材料和方法
1.1 试验材料与试剂
18 胚龄和19 胚龄太行鸡胚毛囊、肝脏均采集于河北农业大学孵化室;Trans2K® Plus DNA Marker、TransFast® Taq DNA Polymerase 为北京全式金公司产品;Super GelRed 和2xES Taq MasterMix(Dye)为保定康为世纪公司产品;pMD™ 19-T Vector Cloning Kit和S1 Nuclease 为大连宝生物公司产品,引物合成及测序由苏州金唯智公司完成。
1.2 RNA 和DNA 提取及cDNA 合成
使用TransZol Up(北京全式金)提取太行鸡毛囊总RNA,EasyPure® Genomic DNA Kit(北京全式金)提取太行鸡肝脏DNA,电泳检测提取DNA 和RNA 的完整性,调整DNA 和RNA 浓度分别为50和1 000 ng/μL; 使用TransScript® One-Step gDNA Removal and cDNA Synthesis SuperMix 试剂盒(北京全式金)将RNA 反转录为cDNA。
1.3 引物设计和PCR 反应
NCBI 下载ev21全长序列(KY235336.1),利用Primer Premier 5.0 设计引物659-S 和659-A 检测样品ev21阴阳性。参考杜小龙[21]研究结果合成477-A 和477-S 引物扩增PRLR和SPEF2的5'共有区域,以半定量PCR 方法明确PRLR和SPEF2基因连接方式。参考张乐超[22]融合基因(fusion)和SPEF2基因检测引物,检测其转录。
PCR 反应体系:2×ES Taq MasterMix(Dye) 5 μL,上下游引物各0.5 μL,DNA 模板1 μL,ddH2O 补齐10 μL。PCR 反应程序:预变性95 ℃ 5 min,变性95 ℃ 30 s,各自退火温度退火30 s,延伸72 ℃ 40 s,35 个循环(除半定量PCR 24 个循环),终延伸72 ℃10 min,4 ℃保存。样品羽型和性别检测参考张秀玲[8]和胡锐颖[23]的检测方法。试验用引物信息见表1。

表1 试验用引物Table 1 Primers used in the experiment
1.4 PRLR 和SPEF2 基因连接方式及共有区域转录方向
1.4.1PRLR和SPEF2基因共有区域拷贝数检测半定量PCR 扩增太行鸡PRLR与SPEF25'共有区域。反应体系为Mix 5 μL,477-S、477-A 各0.3 μL,模板DNA 1 μL,ddH2O 补足10 μL。反应程序:94 ℃预变性5 min,94 ℃变性30 s,60 ℃退火30 s,72 ℃延伸30 s,24 个循环,72 ℃终延伸10 min,4 ℃终止反应。1 %琼脂糖凝胶电泳检测PCR 产物,ImageJ 软件分析目的条带灰度值。
1.4.2PRLR和SPEF2基因共有区域转录方向检测利用477-S 和477-A 特异引物分别对RNA 反转录,得到的cDNA 稀释20 倍。以此cDNA 为模板依照1.4.1 的PCR 条件和体系进行35 个循环的扩增,检测PRLR和SPEF2共有区域转录方向。将目的条带纯化回收与pMDTM19-T 载体连接。构建好的载体进行大肠杆菌转化,菌液送金唯智公司测序,明确转录产物。
1.5 Fusion 基因的转录方向的检测
分别以Fusion-S 和Fusion-A 进行为引物进行特异性反转录,将得到的cDNA 为模板用双引物扩增,1%琼脂糖凝胶检测PCR 结果,检测FUSION基因是否双向转录;将目的片段纯化回收并连接测序,明确扩增产物是否为目标基因序列。
同样进行引物SPEF2-S 和SPEF2-A 以及659-S和659-A 的特异反转录PCR(RT-PCR),检测SPEF2和ev21基因的转录方向,分别作为单向和双向转录对照。
1.6 S1 酶切法检测基因转录方向
以通用引物反转录获得cDNA,将cDNA 产物进行S1 核酸酶酶切,酶切体系:cDNA 1 μg,10×S1 buffer 2 μL,S1 0.06 μL, 灭菌水加到20 μL;酶切温度23 ℃,时间为20 min。另外,取10 μL 酶切产物加入0.5 mL/L EDTA。将cDNA、酶切产物和加EDTA 产物分别为模板进行SPEF2特异性RTPCR。设置3 组通用引物反转录cDNA 的S1 用量和酶切时间:0.06 μL S1,酶切20 min;0.10 μL S1,酶切20 min;0.06 μL S1,酶切30 min,然后以ev21和fusion 引物进行PCR 扩增,分析S1 酶切检测转录方向的效果。
2 结果与分析
2.1 PRLR 和SPEF2 基因连接方式及共有区域转录方向鉴定
2.1.1PRLR和SPEF2基因共有区域拷贝数检测对PRLR与SPEF2共有区域477 bp 片段进行半定量PCR 扩增,1%琼脂糖凝胶检测,结果如图1 所示扩增条带单一清晰,无拖尾现象。纯合慢羽公鸡(7和8 泳道)、杂合慢羽公鸡(1 和2 泳道)、快羽公鸡(3 泳道)、慢羽母鸡(4 泳道)和快羽母鸡(5和6 泳道)的条带亮度存在差异。

图1 PRLR 和SPEF2 基因共有区域半定量PCR 凝胶检测Fig.1 Semi-quantitative PCR gel detection of shared region for PRLR and SPEF2 gene
试验共检测31 只纯合慢羽公鸡、34 只杂合慢羽公鸡、35 只慢羽母鸡、28 只快羽公鸡和33 只快羽母鸡的477 bp 电泳条带灰度值,得到5 种类型鸡的灰度比值依次为2.5∶2.0∶1.6∶1.6∶1(见表2)。
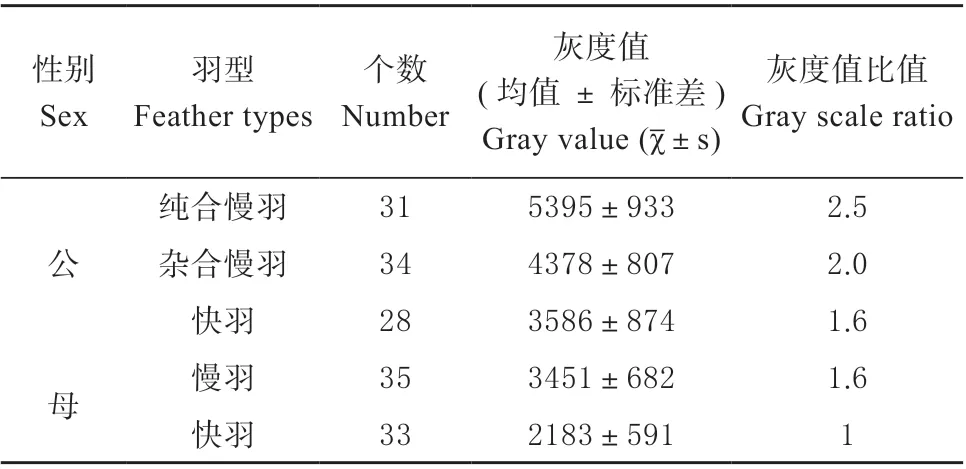
表2 PRLR 和SPEF2 基因共有区域477 bp 灰度值Table 2 Gray value of 477 bp in PRLR and SPEF2 common region
2.1.2PRLR和SPEF2基因共有区域转录方向检测分别以477-S 和477-A 引物进行特异性RT-PCR,结果如图2 所示:泳道1 和2 分别是以477-S 和477-A 特异反转录cDNA 的PCR 产物。回收纯化扩增条带,连接测序,发现477-S 特异反转录cDNA的PCR 产物与目的序列完全一致,而477-A 的PCR主要扩增产物为非特异扩增。
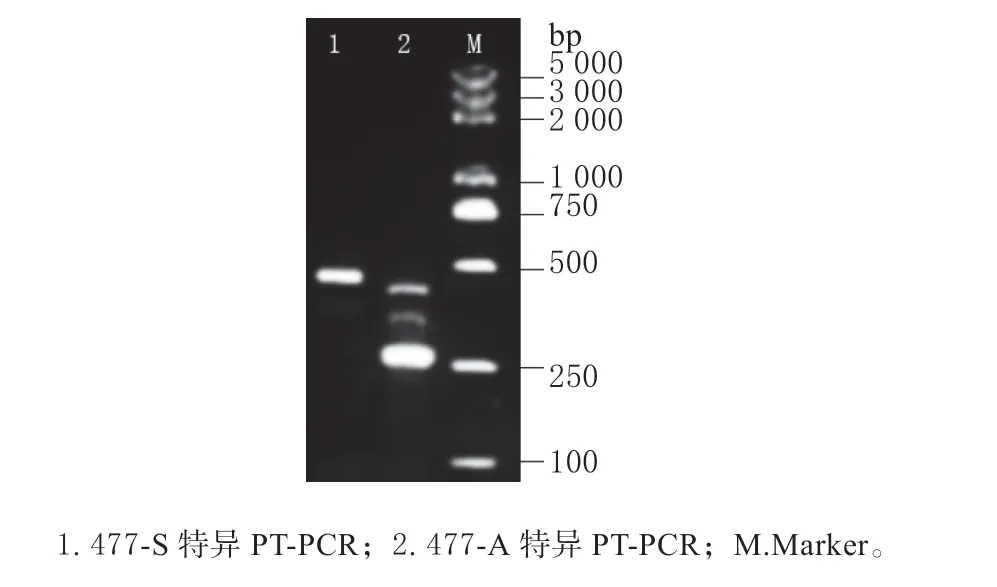
图2 477 bp 特异性反转录的PCR 产物凝胶检测Fig.2 Gel assay of PCR product for 477 bp from specific reverse transcription
2.2 Fusion 基因转录方向的检测
Fusion-S 和Fusion-A 特异反转录融合基因的PCR 产物(图3A)可见清晰单一条带;659-S 和659-A 特异反转录ev21 的PCR 产物(图3B),条带单一,双向均有目的产物;SPEF-S 和SPEF-A 特异反转录SPEF2 产物(图3C),可见2 条单一条带。
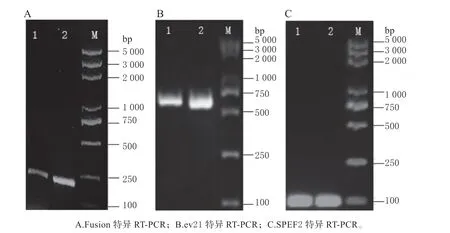
图3 特异反转录的PCR 凝胶检测Fig.3 Gel assays of PCR for specific reverse transcription
对fusion、ev21 和SPEF2的扩增条带克隆测序。fusion测序结果比对(图4)显示,上下游引物各自的特异RT-PCR 扩增获得的片段与目的序列完全一致,说明fusion基因为双向转录。
ev21 的双向特异RT-PCR 产物序列比对发现上下游引物的特异转录产物存在SNP 位点。SPEF2的产物测序发现SPEF-A 的RT-PCR 产物序列与原序列完全匹配,而SPEF-S 的RT-PCR 产物为非特异扩增。

图4 Fusion 特异反转录扩增与原序列比对结果Fig.4 Sequences comparison of specific RT-PCR for Fusion
2.3 S1 酶切产物PCR 结果
将通用引物反转录产物进行S1 酶切后为模板进行SPEF2目的片段扩增,结果见图5A。1、4、7 和10 泳道为模板cDNA 未经处理的扩增,可见目的条带;2、5 和8 泳道为模板经0.06 μL S1 酶切20 min 的扩增,有目的条带;3、6 和9 泳道为模板加EDTA 抑制剂的酶切产物扩增,未见目的条带。
图5B 为ev21的扩增,未经处理cDNA 模板(1泳道)、0.06 μL S1 酶切20 min 模板(2 泳道)和0.06 μL S1 酶切30 min(4 泳道)的ev21 均出现目的条带,而0.10 μL S1 酶切20 min(3 泳道)未出现目的条带。图5C 为Fusion 的扩增,未经处理cDNA 模板(1 泳道)和0.06 μL S1 酶切20 min 模板(2 泳道)出现目的条带,0.10 μL S1 酶切20 min(3 泳道)和0.06 μL S1 酶切30 min(4 泳道)未出现目的条带。
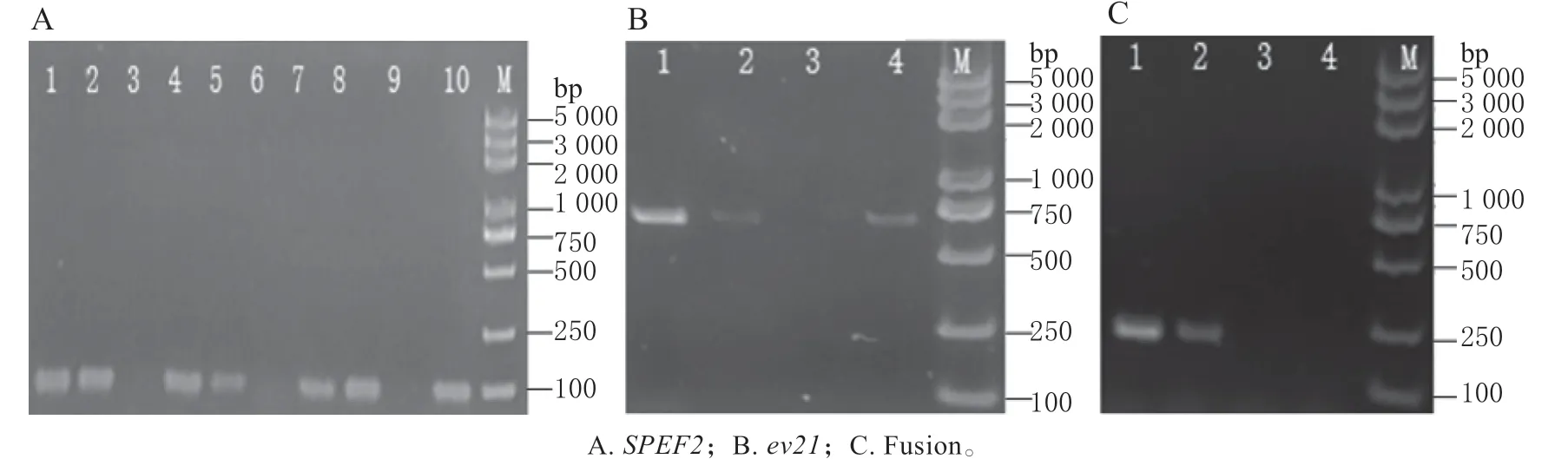
图5 S1 酶切cDNA 模板PCR 扩增凝胶检测Fig.5 Gel detection of PCR of cDNA digested by S1
3 结论与讨论
3.1 PRLR 和SPEF2 基因连接方式及共有区域转录方向
鸡Z 染色体的PRLR与SPEF2基因“头碰头”连接,有477 bp 的共有区域。如果慢羽鸡的dPRLR和dSPEF2重复基因插入不影响PRLR和SPEF2的连接及其与下游基因连接,那么dPRLR和dSPEF2整合在PRLR和SPEF2内部,并且PRLR与dSPEF2以及dPRLR与SPEF2均在5'连接处存在477 bp 共有区域,因此,慢羽鸡Z 染色体上有2 个拷贝的共有区域。公母鸡性染色体分别为ZZ 和ZW 型,所以477 bp 共有区域在纯合慢羽公鸡、杂合慢羽公鸡、慢羽母鸡、快羽公鸡和快羽母鸡基因组中拷贝数分别为4、3、2、2 和1。因此,PCR 检测477 bp 扩增条带灰度值在以上5 种鸡的比例理论上为4∶3 ∶2 ∶2 ∶1,检测结果为2.5 ∶2.0 ∶1.6 ∶1.6 ∶1。虽然与预期比例4∶3∶2∶2∶1 存在差异,PCR 的模板起始浓度、扩增效率、扩增的平台效应以及软件分析的试验误差等都对试验结果产生影响,但试验结果一定程度上证明了预期理论值,可以推断慢羽K 基因中PRLR和dPRLR反向定位在Z 染色体,分别与dSPEF2和SPEF2的5'末端以“头碰头”方式连接。慢羽K 基因分子构造特征的明确为深入挖掘其分子调控机制奠定基础。
试验进一步检测PRLR和SPEF2基因共有区域的转录方向发现,只有477-S 引物特异RT-PCR 成功获得目的片段,而477-A 出现非特异扩增,表明该区域仅在反义链的转录,即PRLR基因有转录,而SPEF2无转录。
3.2 Fusion 基因转录方向
dPPLR由PRLR基因的第1 ~11 外显子以及第12 外显子的558 bp 构成,SPEF2基因的第1 ~5 外显子重复构成dSPEF2[6],dSPEF2和dPPLR的3'末端区域称为融合基因Fusion,该末端连接区域是慢羽K 基因的特异断裂连接点,为慢羽鸡分子检测的靶点。
本试验对fusion、ev21及SPEF2进行特异性RT-PCR,结果显示Fusion 和ev21上下游引物分别反转录得到的cDNA 均可成功扩增出目的条带,且2 个扩增产物与原序列一致,说明fusion和ev21均为双向转录。这与Zhao 等[10]发现fusion具有双向转录产物一致,但尚需RACE 试验进一步明确转录本结构特征。由于双链RNA 可能形成RNA 干扰,即双链RNA 诱发同源mRNA 高效特异性降解[24]。据此猜测fusion 可能影响PRLR和SPEF2的转录,进而影响羽型,但是具体调控机制还需后续试验进行验证。
戴振清[25]发现ev1的反义转录产物lnc-ALVE1-AS1 在禽白血病病毒抗性品系鸡中的表达高于易感品系,与ev1序列高度相似的ev21整合在PRLR基因内含子中,可能存在相似的反向转录产物参与抗禽白血病病毒感染过程。以SPEF-A 进行特异性反转录的cDNA 为模板进行PCR 扩增获得产物与SPEF2原序列完全匹配,而SPEF-S 由于上游引物出现错配导致产生非特异扩增,说明SPEF2为单向转录。通过以上结果发现,特异性反转录因引物设计容易出现假阳性,还需后续测序比对,才能准确鉴定基因的转录方向。
3.3 酶切法检测基因转录方向
S1 核酸酶是单链特异的核酸内切酶,在最适环境下,能将单链核酸或双链核酸中的单链部分降解成为可溶性5'-单核苷酸,但对双链核酸相对不敏感[26]。本试验以3 种方式处理的cDNA 为模板(空白对照、S1 酶切处理和酶切后添加EDTA 抑制剂)扩增SPEF2,证明S1 酶切检测转录方向的可行性。结果显示S1 处理与否均能扩增出目的条带,说明S1 酶切cDNA 并未将单向转录的SPEF2转录本降解,SPEF-S 引物非特异性的扩增产物与特异性产物可能形成部分双链,导致S1 酶切没有降解单向转录的SPEF2转录本。EDTA 处理的cDNA 均未扩增出条带,由于EDTA 螯合了反应体系中的Mg2+,不仅抑制了S1 核酸酶的活性,也抑制了PCR 扩增的Taq酶的活性。调整酶量和酶切时间进行试验,结果显示酶量及酶切时间对试验结果有较大影响,S1核酸酶过量也会水解双链RNA;S1 酶切30 min 造成酶切结果的不稳定性。表明通过S1 核酸酶酶切来判断基因转录方向有很大的局限性。
本试验通过半定量PCR 明确了鸡慢羽K 基因中PRLR和dPRLR反向定位在Z 染色体上,分别与dSPEF2和SPEF2的5' 末端以“头碰头”方式连接;通过特异性反转录证明了PRLR和SPEF2的5'端共有区域只有PRLR发生转录,以及dPRLR和dSPEF2的“尾对尾”3'末端为双向共转录;另外,发现S1 核酸酶酶切法验证基因转录方向存在局限性。本研究为深入探究慢羽鸡K 基因的分子结构特征、挖掘其功能调控提供理论参考。
