气相色谱电子捕获法在检测药品中氯代乙烷类毒性杂质中价值
2021-07-06薛毅博孙红涛王佳佳
薛毅博,孙红涛,王佳佳
驻马店市食品药品检验所药理室,河南 驻马店 463000
药品中的杂质,为无任何疗效的物质,且可能引起患者出现副作用[1]。因此,必须严格控制杂质水平,以确保药品的安全性达到人体用药的要求。根据ICH指南[2],原料药的杂质主要分为有机杂质、无机杂质和残留溶剂三个类别,而在这三类中,遗传毒性杂质安全阈值较低,甚至在低浓度的情况下发生突变,从而可能损伤DNA[3]。Benigin等[4]归纳总结了具有遗传毒性警示结构。但目前,仅有少数文献报道了关于药品中氯代乙烷类化合物含量的检测方法。气相色谱仪电子捕获检测器是一个选择性较好、灵敏度超高的检测器,它只对含卤素、硫、磷等的物质有超强的信号,并且要检测的物质电负性越强,它的检测灵敏度也就越高[5]。SYL930[6]是一种选择性S1P1受体激动剂,由中国医学科学院药物研究所研发,由于它可以明显改善体内的外周血淋巴细胞水平,所以通常用于对类风湿性关节炎患者的治疗,但在合成SYL930的过程中很可能产生氯代乙烷类毒性杂质。因此,本研究建立了一种气相色谱电子捕获法分析方法,同时检测SYL930原料药中可能存在的1,1,2,2-四氯乙烷、1,1-二氯乙烷和1,2-二氯乙烷等3种氯代乙烷类毒性杂质,灵敏度较高,现报告如下。
1 材料
仪器:带有ECD的气相色谱仪(安捷伦科技有限公司),DB-624(30 m×0.53 mm,3.0μm)色谱柱(北京慧安清检测科技有限公司),MSA225S电子天平(赛多利斯科学仪器有限公司)。
药品:1,2-二氯乙烷(GCS,99.6%)、1,1,2,2-四氯乙烷(GCS,99.9%)、1,1-二氯乙烷(GCS,99.5%);N-甲基吡咯烷酮(NMP,色谱纯,99.5%);SYL930原料药(批号:SNT20140402,SNT20140404)。
2 方法及结果
2.1 色谱条件
色谱柱的起始柱温设置为35℃,温度持续4 min以后,以15℃/min的速度快速升到100℃,然后以20℃/min的速度升高温度到230℃,持续20 min。之后采用纯度≥99.99%的高纯氮气,柱的流速设置为4.5 mL/min,分流比设置为5∶1,立即进样,进药量为1μL。
2.2 制备溶液
2.2.1 对照品溶液的制备:精密称量1,2-二氯乙烷100 mg,放入50 m L量瓶中,之后用NMP定容并摇匀得到1,2-二氯乙烷溶液。精密称量1,1,2,2-四氯乙烷10 mg,置于100 mL量瓶中,用NMP定容并充分摇匀后将该溶液定量稀释100倍,制得1,1,2,2-四氯乙烷溶液。精密称量1,1-二氯乙烷100 mg,放到10 mL量瓶后用NMP定容,充分摇匀后制得1,1-二氯乙烷溶液。
2.2.2混合对照品溶液的制备:精密量取上述的1,2-二氯乙烷溶液、1,1-二氯乙烷溶液各1 mL,并精密量取1,1,2,2-四氯乙烷溶液5 mL,置于100 mL量瓶中,之后用NMP定容至刻度并充分摇匀制得混合对照品溶液。精密量取1 mL上述混合对照品溶液放入10 mL量瓶中,然后用NMP定容并充分摇匀后制得混合的对照品溶液。
2.2.3供试品溶液的制备:准确称量200 mg供试品倒入1 mL量瓶中,然后用适量的NMP溶液进行超声溶解处理,之后用NMP定容至刻度并充分摇匀得到供试品溶液。
2.2.4系统适用性溶液的制备:准确称量200 mg供试品放于1 mL量瓶中,精密量取0.1 mL上述混合对照品溶液后倒入到上述量瓶中,然后用NMP定容至刻度并充分摇匀后制得系统适用性溶液。
2.3 溶液稳定性的检测
取适量的上述混合对照品溶液,按照上述“2.1”项的色谱条件,每隔2 h测定一次,共测12次,即24小时,分别记录氯代乙烷类化合物各组分的峰面积,结果显示1,1,2,2-四氯乙烷、1,1-二氯乙烷、1,2-二氯乙烷的RSD值分别为1.8%,0.89%,1.6%,表明在24 h内混合对照品溶液的稳定性良好。
2.4 标准曲线
精密量取上述混合对照品溶液,按照不同比例用NMP定量稀释此溶液,制得不同浓度混合对照品溶液,然后按照“2.1”项色谱条件测定后得到3种化合物的色谱图,峰面积作为纵坐标,各化合物的浓度作为横坐标,采用线性回归分析方法,见表1。

表1 氯代乙烷类化合物的标准曲线
2.5 精密度的测定
分别精密量取上述对照品溶液适量,将每种溶液按照不同比例定量稀释成高、中、低3种不同浓度的溶液,按照“2.1”项色谱条件进行检测分析,并且每种浓度的溶液平行4份,详细记录色谱峰并用峰面积来计算精密度大小,见表2。
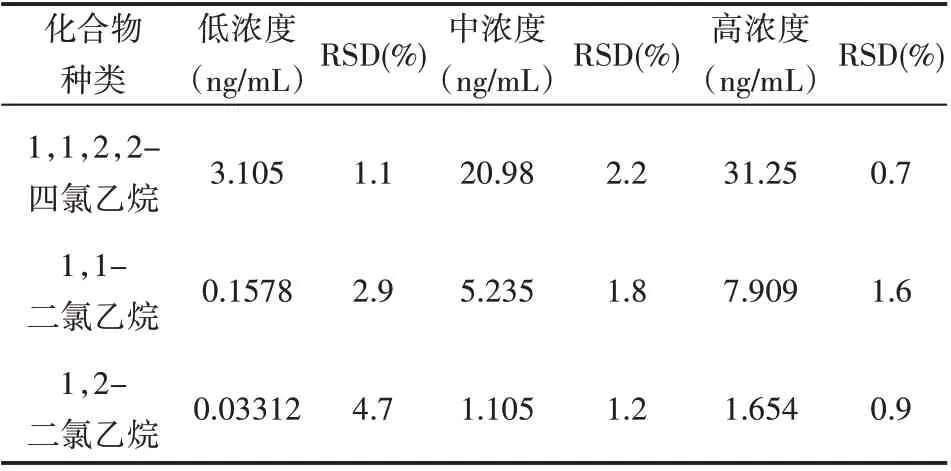
表2 氯代乙烷类化合物的精密度
2.6 最小检测限和最小定量限
取适量的上述混合对照品溶液,用NMP稀释得到适当浓度的溶液,以“2.1”项色谱操作标准进行进样,以信噪比(S/N)作为样品的最小检测浓度标准,以各个组分线性范围的最低浓度表示化合物的最小定量浓度。取适量的氯代乙烷类化合物溶液后定量稀释至3种化合物浓度均为定量限的15倍,得到验证溶液。准确称量供试品200 mg,放入1 mL量瓶后取适量的NMP进行超声溶解处理,加入100μL上述验证溶液,加NMP到刻度处,充分摇匀后按照“2.1”项色谱条件进行含量测定,见表3。
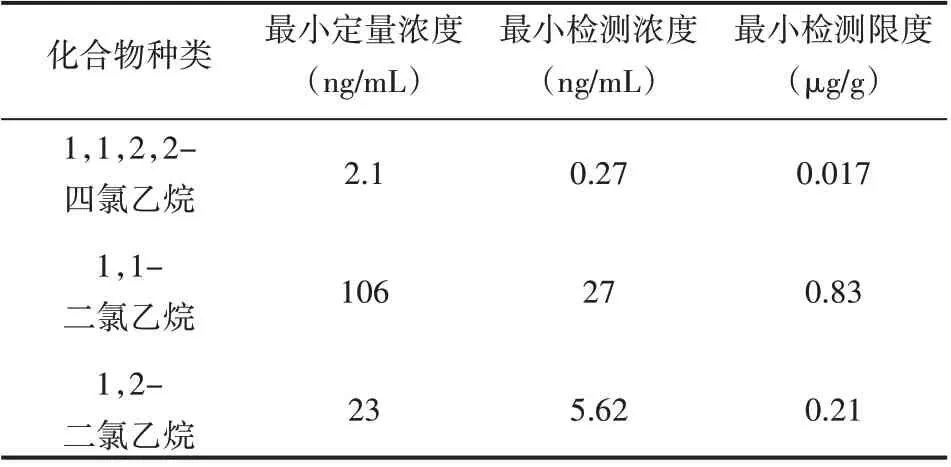
表3 氯代乙烷类化合物的最小检测限和最小定量限
2.7 含量的检测
精密称取200 mg供试品放到1mL量瓶中,平行实验3份,采取适量的NMP进行超声溶解处理后,再用NMP稀释至刻度并充分摇匀,按照“2.1”项色谱条件进行检测分析,详细记录色谱图,采用外标法来计算峰面积,以此来表示含量。结果发现,在原料药SYL930中未检测出含有1,1,2,2-四氯乙烷、1,1-二氯乙烷和1,2-二氯乙烷等毒性杂质。
3 讨论
本研究建立了SYL930原料药中1,1,2,2-四氯乙烷、1,1-二氯乙烷和1,2-二氯乙烷等氯代乙烷类化合物的气相色谱电子捕获法检测,优化了色谱的检测条件。
本研究采用DB-624色谱柱,因为该色谱柱峰形对称且比较窄,无拖尾现象[7],可以实现对氯代乙烷类化合物的有效分离;本研究采用ECD检测器,因为要分离的化合物含有氯原子,在该检测器上比较灵敏。本研究采用以15℃/min升温,因为当以15℃/min升温时,氯代乙烷类化合物的各组分形成的峰形较窄,并且可以实现氯代乙烷类化合物与NMP和供试品中的杂质峰分离完全[8];本研究对于溶液的制备,采用准确称量、精密量取的操作方法,用NMP定容至刻度,目的是使检测结果更可靠、更具有说服性;本研究采用5:1的分流比,因为进药量过大会引起色谱的污染且还会引起基线噪声的增加[9]。本研究发现1,1,2,2-四氯乙烷在2.096~42.56 ng/mL、1,1-二氯乙烷在0.1078~10.69μg/mL、1,2-二氯乙烷在0.02136~2.136μg/m L范围内,线性关系均良好,且相关系数均大于0.999;上述三种化合物的在低、中、高浓度的RSD值0.9%~4.7%,说明该方法精密度良好;1,1-二氯乙烷的最小检测浓度为27 ng/m L、最小检测限度为0.83μg/g,相比于其他两种化合物均较高,检测结果发现原料药中的3种氯代乙烷类化合物最小定量限均<1 ppm,且信噪比>10,符合检测基因毒性杂质的需要[10],可十分有效地检测原料药中的各组分,从而可以检测药物的有毒杂质。
综上所述,故采用气相色谱电子捕获法、用DB-624色谱柱建立的方法检测氯代乙烷类毒性杂质专属性良好、灵敏度较高、准确度和精密度较好,值得推广应用。
