竹柏叶中黄酮类成分的分离与鉴定
2021-07-03郭婕,王欢,张祁,宋科
郭 婕, 王 欢, 张 祁, 宋 科
(吉首大学 林产化工工程湖南省重点实验室,湖南 张家界 427000)
竹柏(Nageianagi(Thunberg) Kuntze)是罗汉松科竹柏属植物,多分布于长江以南各省区[1],是常见的园林绿化用树种。竹柏中含有黄酮类、去甲基二萜双内酯类、挥发油等多种类型的活性化合物[2-5],其中双黄酮类化合物广泛存在于松柏纲和银杏纲等植物中,是由两分子相同或不同类型的黄酮及其衍生物为单元聚合而成的二聚物,在抗肿瘤和治疗心血管疾病等方面活性显著[6-8]。穗花杉双黄酮是一种在松柏纲植物中分布广泛的双黄酮类化合物,以卷柏科卷柏属(Selaginella)植物中含量最高,具有抗炎、抗氧化和抑菌等生理功能,在缓解动物应激、减少炎症损伤、提高动物抵抗致病菌的能力等方面具有巨大潜力[9-10],已在医药、保健等领域有所应用[11-12],有望在畜禽生产中得以开发利用。目前,关于大孔树脂分离纯化药用植物中总黄酮的工艺研究[13-15]屡见报道,但有关竹柏总黄酮和其中穗花杉双黄酮的分离纯化工艺却鲜有报道。本课题组前期对竹柏枝叶提取物中的总黄酮进行定量研究[16],结果显示竹柏枝叶粗提物中总黄酮的质量分数可达13.6%,且各部位有着显著的差异,叶15.5%>茎10.4%>根8.5%>种子0.15%。在此基础上,本研究比较了大孔吸附树脂和聚酰胺(PLA)树脂对竹柏总黄酮的吸附-解吸性能,确立AB-8大孔吸附树脂对竹柏总黄酮的分离富集工艺,并进一步对竹柏总黄酮进行纯化,从竹柏叶正丁醇萃取物中分离得到8个单体化合物,通过各种波谱手段鉴定化合物的结构,以期为竹柏提取物的组成成分和生物活性研究提供参考,为寻找双黄酮的替代药源提供理论依据。
1 实 验
1.1 材料、试剂及仪器
竹柏叶于2014年12月采自湖南省张家界国家森林公园,由吉首大学谷伏安副教授鉴定为竹柏(Nageianagi(Thunberg) Kuntze),50 ℃烘干,粉碎后放入干燥器内备用。芦丁标准品(批号:Y10A6S1,纯度≥98%),上海源叶生物科技有限公司;穗花杉双黄酮标准品(批号:MUST-15012505,纯度≥99.05%),北京世纪奥科生物技术有限公司;色谱甲醇试剂,天津康科德科技有限公司;乙醇(体积分数75%)、石油醚、氯仿、丙酮、正丁醇、甲醇、氢氧化钠、亚硝酸钠和硝酸铝等,均为市售分析纯。
ZF-I型三用紫外(UV)分析仪,日本日立公司;Nicolet Is 10型傅里叶变换红外光谱(FT-IR)仪,DSQ Ⅱ EI质谱(MS)仪,美国Thermo Scientific公司;Agilent 1260型高效液相色谱(HPLC)仪,美国安捷伦科技有限公司;Bruker AVANCE Ⅲ HD 500型核磁共振波谱(NMR)仪(TMS为内标)。
各种规格的玻璃层析柱,北京欣维尔玻璃仪器有限公司;AB-8大孔吸附树脂、D101大孔吸附树脂、聚酰胺树脂-1号(PLA-1)和聚酰胺树脂-2号(PLA-2),郑州勤实科技有限公司;硅胶(150~180 μm、48~75 μm和38~48 μm)和GF254薄层层析硅胶,青岛海洋化工厂;凝胶SephadexLH-20(40~70 μm),瑞典Amersham Pharmacia Biotech公司;反相填充材料YMC*GEL (ODS-A-HG,S-50 μm),日本YMC株式会社。
1.2 竹柏叶总黄酮的提取
1.2.1竹柏叶提取物的制备 取干燥的竹柏叶5.0 kg,粉碎,用体积分数70%乙醇超声波提取5次,合并提取液,减压浓缩得到浸膏。
1.2.2竹柏叶总黄酮质量浓度的测定 称取干燥的竹柏叶40 g,加入1 000 mL体积分数为70%的乙醇于60 ℃水浴中超声波提取30 min,提取3次,合并提取液,减压浓缩至干燥。然后用蒸馏水溶解、过滤后,配制成不同质量浓度的样品溶液,备用。精确称取经105 ℃干燥至质量恒定的芦丁标准对照品21.2 mg,用适量的体积分数为70%的甲醇溶解,加水定容至50 mL容量瓶中,配制成0.424 g/L芦丁标准品母液。分别量取0、 0.2、 0.4、 0.6、 0.8、 1.0和1.2 mL芦丁标准品母液于10 mL容量瓶中,加入2.0 mL无水甲醇,摇匀,各加入5%亚硝酸钠溶液0.4 mL,摇匀后静置6 min,再分别加入10%硝酸铝溶液0.4 mL,摇匀后静置6 min,再分别加入4 mL 1 mol/mL氢氧化钠溶液,摇匀后静置15 min,加水定容至刻度,摇匀后置于比色皿中,于506 nm波长下测定吸光度,以芦丁标准对照品溶液质量浓度(C,g/L)为横坐标,吸光度(A)为纵坐标,得到线性回归方程:A=11.0C+0.024 8(R2=0.999 1),线性范围0.013~0.076 g/L[16]。
准确量取一定量的样品溶液,测定吸光度,根据标准曲线计算竹柏叶总黄酮的质量浓度。
1.2.3穗花杉双黄酮质量浓度的测定 精确称取穗花杉双黄酮标准对照品2.1 mg,用5.0 mL体积分数为70%的甲醇溶解,定容至10 mL的容量瓶中,配制成质量浓度为0.21 g/L穗花杉双黄酮标准品母液,过0.45 μm滤膜,备用。分别量取0.1、 0.2、 0.4、 0.8、 1.0、 2.0和4.0 mL穗花杉双黄酮标准品母液于10 mL的容量瓶中,用体积分数70%的甲醇定容至刻度线处,摇匀。采用HPLC法进行检测,色谱条件为:色谱柱Kromasil C18柱(4.6×250 mm, 5 μm),检测波长335 nm,柱温箱35 ℃,流速1.0 mL/min;进样量10 μL;流动相使用0.2%磷酸缓冲溶液(A)/甲醇(B)进行梯度洗脱(0~15 min,40% A;15~30 min,15% A),记录标准品溶液的峰面积,绘制穗花杉双黄酮标准曲线,采用外标法计算样品中穗花杉双黄酮的质量分数。以穗花杉双黄酮标准对照品溶液质量浓度(X,g/L)为横坐标,峰面积(Y)为纵坐标,得到线性回归方程为:Y=678 23X-31.297(R2=0.999 5),线性范围0.002 1~0.084 0 g/L。
1.3 树脂的筛选
1.3.1静态吸附-解吸实验
1.3.1.1吸附率及解吸率的计算 分别称取经预处理的4种型号(聚酰胺1号、聚酰胺2号、D101,AB-8)的树脂各1.0 g(干质量),置于50 mL的锥形瓶中,分别加入总黄酮质量浓度为8.78 g/L竹柏乙醇提取液(1.2.2节中制备)20 mL,密封,置于25 ℃恒温水浴振荡器中振荡24 h,静置后过滤,测定溶液中总黄酮和穗花杉双黄酮的质量浓度,并按照式(1)~(2)分别计算吸附量和吸附率。
(1)
(2)
式中:qa—吸附量,mg/g;Ea—吸附率,%;C0—吸附前初始质量浓度,g/L;Ce—吸附后的质量浓度,g/L;V—吸附液体积,mL;m—树脂质量,g。
将吸附饱和的4种型号树脂,用一定量的蒸馏水冲洗,抽干,置于50 mL的锥形瓶中,分别加入20 mL 体积分数70%的乙醇溶液,密封,置于25 ℃恒温水浴振荡器中振荡24 h,静置后过滤,测定滤液中总黄酮和穗花杉双黄酮的质量浓度,并按照式(3)~(4)分别计算解吸量和解吸率。
(3)
(4)
式中:qd—解吸量,mg/g;Ed—解吸率,%;Cd—解吸液的质量浓度,g/L;V—解吸液体积,mL;m—树脂质量,g。
1.3.1.2上样液质量浓度对吸附效果的影响 分别称取经预处理的AB-8大孔吸附树脂、聚酰胺树脂各1.0 g(干质量),置于50 mL的锥形瓶中,分别加入总黄酮质量浓度为0.5、 1.0、 1.5、 3.0和4.0 g/L (其中穗花杉双黄酮质量浓度7.15、 12.56、 13.90、 32.36和43.97 mg/L)的样品溶液20 mL,密封,置于25 ℃恒温水浴振荡器中振荡24 h,静置,过滤,考察不同上样溶液质量浓度对竹柏总黄酮吸附量和吸附率的影响。
1.3.1.3洗脱剂体积分数对解吸效果的影响 将吸附饱和的AB-8大孔吸附树脂和聚酰胺树脂,用一定量的蒸馏水冲洗除去表面残留的溶液,抽干,置于50 mL的锥形瓶中,分别加入20 mL体积分数为50%、 60%、 70%、 80%和90%的乙醇溶液,密封,置于25 ℃恒温水浴振荡器中振荡24 h,静置后过滤,考察不同体积分数的洗脱剂对竹柏总黄酮解吸量和解吸率的影响。
1.3.2动态吸附-解吸实验
1.3.2.1吸附流速对吸附效果的影响 按照1.3.1节的条件,将200 mL样品溶液以22、 44 和88 mL/h的吸附流速上样,每10 mL收集1管流出液,定容,分别测定流出液中总黄酮和穗花杉双黄酮的质量浓度,考察不同吸附流速对吸附效果的影响。
1.3.2.2上样量对吸附效果的影响 按照1.3.1节的条件,将树脂湿法装柱后,取一定量的样品溶液以22 mL/h的流速吸附上样,每10 mL收集1管流出液,定容,分别测定流出液中总黄酮和穗花杉双黄酮的质量浓度。
1.3.2.3洗脱流速对解吸效果的影响 将吸附饱和的AB-8大孔吸附树脂柱,用蒸馏水冲洗去除表面残留的溶液,之后将洗脱剂分别以22、 44和88 mL/h的流速解吸,每10 mL收集1管洗脱液,定容,分别测定洗脱液中总黄酮和穗花杉双黄酮的质量浓度。
1.3.2.4洗脱剂用量对解吸效果的影响 按前述条件将树脂柱吸附至饱和,蒸馏水冲洗去除表面残留的溶液,之后用一定量的洗脱剂以44和88 mL/h的流速洗脱,每10 mL收集1管洗脱液,定容,分别测定洗脱液中总黄酮和穗花杉双黄酮的质量浓度。
1.4 竹柏叶黄酮化合物的纯化
将竹柏叶提取物的浸膏加水制成悬浮液后,依次用石油醚、氯仿和正丁醇萃取。得到石油醚萃取部分(20 g),氯仿萃取部分(30 g),正丁醇萃取部分(200 g)。将竹柏叶正丁醇萃取物用水溶解、过滤后,通过AB-8大孔吸附树脂分离,乙醇/水(体积比0 ∶1~1 ∶0)进行梯度洗脱,收集洗脱液,经硅胶薄层板点样检测划分得到6部分:Fr.1(20.5 g),Fr.2(26.6 g),Fr.3(36.1 g),Fr.4(42.3 g),Fr.5(30.7 g),Fr.6(19.5 g)。Fr.2组分浓缩后,采用硅胶柱色谱,用氯仿/甲醇梯度洗脱(体积比0 ∶1~1 ∶0),收集洗脱液流分。经薄层层析跟踪合并从Fr.2组分中得到4个洗脱部分(Fr.2.1~Fr.2.4)。然后将Fr.2.3用甲醇/水溶液梯度洗脱(体积比7 ∶3~1 ∶1),分别经过聚酰胺柱层析、凝胶柱层析、ODS反相柱色谱和重结晶反复分离纯化,收集流分得到化合物1(25 mg)和化合物3(5 mg)。Fr.3组分浓缩后,采用硅胶柱色谱,用氯仿/甲醇溶液梯度洗脱,收集洗脱液流分。经薄层层析跟踪合并从Fr.3组分中得到5个洗脱部分(Fr.3.1~Fr.3.5)。然后将Fr.3.4用甲醇/水溶液梯度洗脱,分别经过ODS反相柱色谱、凝胶柱层析、重结晶和制备薄层反复分离纯化,得到化合物2(50 mg)、化合物4(13 mg)和化合物6(4 mg)。Fr.4组分浓缩后,采用硅胶柱色谱,用氯仿/甲醇溶液梯度洗脱,收集洗脱液流分。经薄层层析跟踪合并从Fr.4组分中得到4个洗脱部分(Fr.4.1~Fr.4.4)。然后将Fr.4.2用甲醇/水溶液梯度洗脱,分别经过制备薄层、重结晶和ODS反相柱色谱反复分离纯化,得到化合物5(15 mg)、化合物7(6 mg)和化合物8(33 mg)。
2 结果与讨论
2.1 树脂的筛选
由表1可知,聚酰胺树脂的吸附率明显高于大孔吸附树脂,而大孔吸附树脂解吸率明显优于聚酰胺树脂。因此,选取吸附率相对较高的聚酰胺树脂(PLA-2)和解吸率较高的AB-8大孔吸附树脂进行后续静态吸附-解吸实验研究。

表1 不同树脂的静态吸附-解吸性能
2.2 树脂的静态吸附-解吸
2.2.1上样液质量浓度的影响 从表2可知,在一定浓度范围内,随着上样液质量浓度的不断增加,两种树脂对竹柏黄酮的吸附率均随之增大,当上样液质量浓度为1.5 g/L时,对竹柏黄酮的吸附率达最大值分别为94.88%和90.13%。当上样液质量浓度为3.0 g/L时,穗花杉双黄酮的吸附率达到最大,分别为98.57%和94.68%。因此,为了从竹柏中获取更多的黄酮类化合物,选取上样液质量浓度为1.5~3.0 g/L较为适宜。

表2 不同质量浓度的溶液对吸附率的影响
2.2.2洗脱剂乙醇体积分数的影响 由表3可知,两种树脂对穗花杉双黄酮的解吸率随着洗脱剂乙醇体积分数的增加而逐渐变大,当洗脱剂体积分数为80%时,继续增大洗脱剂体积分数,对竹柏黄酮的洗脱率减小,因此选取体积分数为80%的乙醇作为洗脱剂较好。

表3 不同浓度洗脱剂对解吸率的影响
2.3 树脂的动态吸附-解吸
2.3.1上样流速对吸附效果的影响 如图1可知,在上样量200 mL时,随着上样流速的加快,总黄酮泄露速度加快,分析原因可能是上样流速过快导致树脂与样品溶液接触时间较短,不利于吸附。因此,选取上样流速22 mL/h较适宜。
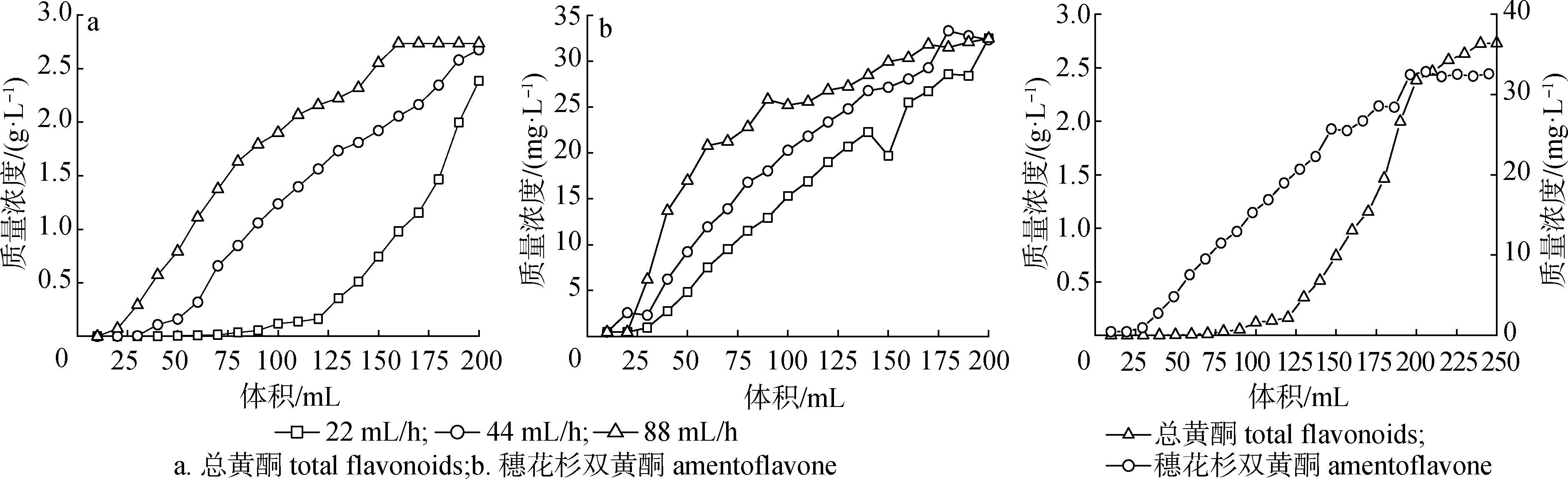
图1 不同上样流速对吸附效果的影响
2.3.2上样量对吸附效果的影响 由图2可知,随着上样量的逐渐增加,流出液中总黄酮的质量浓度增加,树脂吸附效果逐渐下降。当上样量少于200 mL时,虽然流出液中总黄酮和穗花杉双黄酮的质量浓度均明显增加,但是大部分的总黄酮仍然能被树脂吸附。继续加大上样量至220 mL时,流出液中总黄酮质量浓度接近上样液质量浓度,树脂吸附能力明显降低,表明树脂已接近吸附饱和。因此,为减少样液中总黄酮大量泄露使纯化得率降低,选取220 mL为最佳上样量。
2.3.3洗脱流速对解吸效果的影响 如图3可知,在洗脱剂200 mL时,随着洗脱流速的加快,树脂与洗脱剂接触时间较短不利于洗脱,导致解吸率降低。但是,洗脱流速为44 mL/h时与流速为22 mL/h时,洗脱效果相差较小。因此,为了提高工作效率,选取洗脱流速为44 mL/h较适宜。
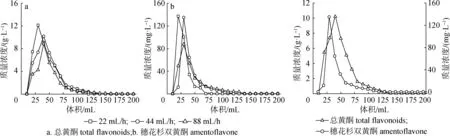
图3 不同洗脱流速对解吸效果的影响
2.3.4洗脱剂用量对解吸效果的影响 由图4可以看出,随着洗脱剂用量的逐渐增加,洗脱液中总黄酮的质量浓度增加。当洗脱剂用量达到140 mL时,洗脱液中几乎检测不到总黄酮和穗花杉双黄酮,表明此时已接近洗脱极限。因此,为减少溶剂的浪费,选取140 mL为最佳洗脱剂用量。
2.3.5验证试验 综合静态吸附-解吸及动态吸附-解吸实验结果,确立了竹柏总黄酮的最佳纯化工艺条件为:采用AB-8大孔吸附树脂以22 mL/h的流速吸附质量浓度为3.0 g/L的竹柏粗提液220 mL,达到吸附平衡后,用140 mL体积分数为80%的乙醇以44 mL/h的流速进行洗脱,测得在该条件下纯化得到的样品中总黄酮平均质量分数为52.8%,穗花杉双黄酮的平均质量分数为2.37%。
2.4 单体化合物结构表征
化合物1,浅褐色结晶,分子式为:C15H14O6,经薄层色谱展开,置紫外灯254 nm下观察,有紫外吸收,喷显色剂(5%浓硫酸-乙醇溶液)加热后呈黄棕色斑点,熔点为174~175 ℃。1H NMR(500 MHz, DMSO-d6):δ4.5(1H,d,J=7.5 Hz, H-2),3.8(1H, m, H-3),2.7(1H, dd,J=4.6 Hz, 16.3 Hz, H- 4a),2.4(1H, dd,J=8.1 Hz, 16.2 Hz, H- 4b),5.7(1H, d,J=2.2 Hz, H- 6),5.7(1H, d,J=2.2 Hz, H-8),6.7(1H, d,J=1.9 Hz, H-2′),6.6(1H, d,J=8.1 Hz, H-5′),6.6(1H, dd,J=1.9 Hz, 8.1 Hz, H- 6′);13C NMR(125 MHz, DMSO-d6):δ81.0(C-2),66.3(C-3),28.2(C- 4),156.3(C-5),95.2(C- 6),155.8(C-7),94.1(C-8),156.6(C-9),99.1(C-10),130.7(C-1′),114.8(C-2′),144.9(C-3′),144.5(C- 4′),115.1(C-5′),118.4(C- 6′)。综合以上理化常数和波谱数据,且化合物1与儿茶素标准品共薄层多体系展开,呈现单一斑点,并与文献[17~18]对照,确定该化合物为儿茶素(catechin)。
化合物2,黄色粉末,分子式为:C15H10O5,经薄层色谱展开,置紫外灯254 nm下观察,有紫外吸收,喷显色剂(5%浓硫酸-乙醇溶液)加热后呈黄色斑点,于365 nm下呈现亮黄色荧光。1H NMR(500 MHz, DMSO-d6):δ7.9(2H, d,J=8.8 Hz, H-2′, H- 6′),6.9(2H, d,J=8.8 Hz, H-3′, H-5′),6.7(1H, s, H-3),6.0(1H, s, H- 6),6.3(1H, s, H-8),12.9(1H, br.s, 5-OH);13C NMR(125 MHz, DMSO-d6):δ163.1(C-2),102.5(C-3),181.1(C- 4),161.3(C-5),99.6(C- 6),163.6(C-7),94.4(C-8),157.5(C-9),105.2(C-10),121.1(C-1′),128.2(C-2′, C- 6′),116.0(C-3′, C-5′),δ162.4(C- 4′)。综合以上波谱数据,推断该化合物为5,7,4′-三羟基黄酮,与文献[19~20]报道芹菜素的数据一致,因此,确定该化合物为芹菜素(apigenin)。
化合物3,白色结晶,分子式为:C15H14O6,经薄层色谱展开,置紫外灯254 nm下观察,有紫外吸收,喷显色剂(5%浓硫酸-乙醇溶液)加热后呈黄棕色斑点,熔点为244~245 ℃。1H NMR(500 MHz, DMSO-d6):δ4.7(1H, br.s, H-2),4.0(1H, m, H-3),2.7(1H, dd,J=5.3 Hz, 16.4 Hz, H- 4a),2.5(1H, dd,J=8.1 Hz, 16.1 Hz, H- 4b),5.7(1H, d,J=2.2 Hz, H- 6),5.9(1H, d,J=2.1 Hz, H-8),6.9(1H, d,J=1.5 Hz, H-2′),6.7(1H, d,J=8.0 Hz, H-5′),6.6(1H, dd,J=1.7 Hz, 8.2 Hz, H- 6′);13C NMR(125 MHz, DMSO-d6):δ78.1(C-2),65.0(C-3),27.9(C- 4),156.2(C-5),95.1(C- 6),155.4(C-7),93.9(C-8),156.5(C-9),98.5(C-10),130.6(C-1′),114.6(C-2′),144.5(C-3′),144.5(C- 4′),115.0(C-5′),118.0(C- 6′)。综合以上理化常数和波谱数据,并与文献[18, 21]对照,确定该化合物为表儿茶素(epi-catechin)。
化合物4,浅黄色粉末,分子式为:C21H20O10,经薄层色谱展开,置紫外灯254 nm下观察,有紫外吸收,喷显色剂(5%浓硫酸-乙醇溶液)加热后呈黄色斑点,于365 nm下呈现亮黄色荧光。1H NMR(500 MHz, DMSO-d6):δ8.0(2H, d,J=8.2 Hz, H-2′, H- 6′),6.9(2H, d,J=8.2 Hz, H-3′, H-5′),6.8(1H, s, H-3),6.3(1H, s, H- 6),13.2(1H, br.s, 5-OH),葡萄糖的端基氢为δ4.7(1H, d,J=9.8 Hz, H-1″),3.2~4.0(6H, m);13C NMR(125 MHz, DMSO-d6):δ164.0(C-2),102.5(C-3),182.1(C- 4),160.4(C-5),98.2(C- 6),161.2(C-7),104.6(C-8),156.1(C-9),104.0(C-10),121.6(C-1′),129.0(C-2′, C- 6′),115.9(C-3′, C- 5′ ),162.8(C- 4′),葡萄糖:δ73.4(C-1″),70.9(C-2″),78.7(C-3″),70.6(C- 4″),81.9(C-5″),61.3(C- 6″)。1H NMR谱与化合物2相比,多了δ4.7(1H, d,J=9.8 Hz)葡萄糖的端基质子,推测该化合物为黄酮碳苷类化合物,同时13C NMR(125MHz, DMSO-d6)中δ104.7也证实为黄酮碳苷C-8的特征信号,δ98.2为C- 6的特征信号;糖端基碳δ73.4,与黄酮氧苷中糖端基碳δ98~112有明显的差别,根据δ73.4,70.9,78.7,70.8,81.9,61.3的一组碳信号,推测糖基为未被取代的β-D-葡萄糖。综合以上波谱数据,并与文献[22~23]对照,确定该化合物为芹菜素-8-C-β-D-葡萄糖,即牡荆苷(vitexin)。
化合物5,黄色粉末,分子式为:C27H30O14,经薄层色谱展开,置紫外灯254 nm下观察,有紫外吸收,喷显色剂(5%浓硫酸-乙醇溶液)加热后呈黄色斑点,于365 nm下呈现亮黄色荧光。1H NMR(500 MHz, DMSO-d6):δ8.1(2H, d,J=8.8 Hz, H-2′, H- 6′),7.0(2H, d,J=8.7 Hz, H-3′, H-5′),6.8(1H, s, H-3),6.2(1H, s, H- 6),13.1(1H, br.s, 5-OH),葡萄糖:δ4.8(1H, d,J=10.1 Hz, H-1″),3.3(1H, m),3.4(2H, m),3.5(1H, m),3.8(1H, m),4.0(1H, m),鼠李糖:δ5.0(1H, s, H-1‴),2.0~4.0(4H, m),0.5(3H, d,J=6.2 Hz);13C NMR(125 MHz, DMSO-d6):δ164.0(C-2),103.7(C-3), 182.0(C- 4),160.7(C-5),98.1(C- 6),162.3(C-7),105.8(C-8),155.8(C-9),104.5(C-10),121.6(C-1′),129.0(C-2′, 6′),115.9(C-3′, 5′),161.2(C- 4′),葡萄糖:δ71.7(C-1″),75.0(C-2″),78.3(C-3″),70.3(C- 4″),80.9(C-5″),61.2(C- 6″),鼠李糖:δ100.3(C-1‴),70.4(C-2‴),70.1(C-3‴),71.5(C- 4‴),68.2(C-5‴),17.7(C- 6‴)。1H NMR谱显示该化合物母体结构为牡荆苷(化合物4)。根据1H NMR谱中出现的甲基的质子信号δ0.5(3H, d,J=6.2 Hz)和糖的端基氢信号δ5.0(1H, s, H-1‴),以及13C NMR谱中一组信号δ100.3,70.4,70.1,71.5,68.2,17.7,推测该化合物中还连有α-L-鼠李糖。综合以上波谱数据,并与文献[20,24]对照,确定该化合物为牡荆苷-2″-O-α-L-鼠李糖,即牡荆苷鼠李糖苷(vitexin rhamnoside)。
化合物6,黄色粉末,分子式为:C30H18O10,经薄层色谱展开,置紫外灯254 nm下观察,有紫外吸收,喷显色剂(5%浓硫酸-乙醇溶液)加热后呈黄色斑点,于365 nm下呈现亮黄色荧光。1H NMR(500 MHz, DMSO-d6):δ6.8(1H, s, H-3),6.5(1H, d,J=2.1 Hz, H- 6),6.2(1H, d,J=2.1 Hz, H-8),8.0(1H, br.s, H-2′),7.16(1H, d,J=8.4 Hz, H-5′),8.0(1H, d,J=2.4 Hz, 8.5 Hz, H- 6′),6.8(1H, s, H-3″),6.4(1H, s, H- 6″),7.6(2H, d,J=8.8 Hz, H-2‴, H- 6‴),6.7(2H, d,J=8.9 Hz, H-3‴, H-5‴),13.1(1H, br.s, 5-OH),13.0(1H, br.s, 5″-OH),10.8(1H, br.s, 7-OH), 10.3(1H, br.s, 7″-OH);13C NMR(125 MHz, DMSO-d6):δ164.1(C-2),103.0(C-3),182.1(C- 4),161.9(C-5),98.8(C- 6),163.7(C-7),94.0(C-8),157.4(C-9),104.0(C-10),121.4(C-1′),131.4(C-2′),116.1(C-3′),160.5(C- 4′),119.9(C-5′),127.8(C- 6′),163.8(C-2″),102.6(C-3″),181.7(C- 4″),161.4(C-5″),98.6(C- 6″),159.5(C-7″),103.6(C-8″),154.5(C-9″),103.7(C-10″),120.9(C-1‴),128.2(C-2‴, C- 6‴),115.7(C-3‴, C-5‴),161.0(C- 4‴)。综合以上理化性质和波谱数据和标准品共薄层检测,并与文献[9,25]对照,且化合物6与穗花杉双黄酮标准品共薄层多体系展开,呈现单一斑点,进一步确定该化合物为穗花杉双黄酮(amentoflavone)。
化合物7,淡黄色粉末,分子式为:C31H20O10,经薄层色谱展开,置紫外灯254 nm下观察,有紫外吸收,喷显色剂(5%浓硫酸-乙醇溶液)加热1min后呈黄色斑点。1H NMR(500MHz, DMSO-d6):δ6.8(1H, s, H-3), 6.2(1H, d,J=2.0 Hz, H- 6), 6.3(1H, d,J=1.6 Hz, H-8), 8.2(1H, d,J=2.1 Hz, H-2′), 7.0(1H, d,J=8.7 Hz, H-5′), 7.9(1H, dd,J=2.3, 8.6 Hz, H- 6′), 6.9(1H, s, H-3″), 6.2(1H, s, H- 6″), 7.8(2H, d,J=8.9 Hz, H-2‴, 6‴), 6.8(2H, d,J=7.4 Hz, H-3‴, 5‴), 13.1(1H, br.s, 5-OH), 13.0(1H, br.s, 5″-OH), 3.67(3H, s, 4‴-OMe);13C NMR(125 MHz, DMSO-d6):δ164.0(C-2), 102.6(C-3), 181.7(C- 4), 161.4(C-5), 98.8(C- 6), 164.0(C-7), 94.0(C-8), 157.3(C-9), 103.6(C-10), 119.3(C-1′), 131.4(C-2′), 121.9(C-3′), 162.7(C- 4′), 118.0(C-5′), 127.1(C- 6′), 164.3(C-2″), 103.1(C-3″), 181.8(C- 4″), 160.5(C-5″), 102.5(C- 6″), 162.6(C-7″), 100.4(C-8″), 154.7(C-9″), 105.6(C-10″), 123.2(C-1‴), 128.0(C-2‴,C- 6‴), 114.3(C-3‴,C-5‴), 162.0(C- 4‴), 55.3(4‴-OMe)。从HMBC谱可知甲氧基上的氢信号δ3.7(3H, s)与δ161.9处C- 4‴相连,综合以上波谱数据,并与文献[17,26]对照,确定该化合物为罗汉松双黄酮A(podocarpus flavone A)。
化合物8,淡黄色粉末,分子式为:C31H20O10,经薄层色谱展开,置紫外灯254 nm下观察,有紫外吸收,喷显色剂(5%浓硫酸-乙醇溶液)加热1 min后呈黄色斑点。1H NMR(400 MHz, DMSO-d6):δ6.9(1H, s, H-3), 6.2(1H, d,J=2.8 Hz, H- 6), 6.5(1H, d,J=2.0 Hz, H-8), 8.2(1H, d,J=2.8 Hz, H-2′), 7.2(1H, d,J=8.5 Hz, H-5′), 8.0(1H, dd,J=2.8, 8.8 Hz, H- 6′), 6.8(1H, s, H-3″), 6.4(1H, s, H- 6″), 7.7(2H, d,J=8.9 Hz, H-2‴,6‴), 6.9(2H, d,J=8.8 Hz, H-3‴,5‴), 3.8(3H, s, 4′-OMe), 13.1(1H, s, 5-OH), 10.8(1H, s, 7-OH), 13.0(1H, s, 5″-OH), 10.4(1H, s, 7″-OH);13C NMR(100 MHz, DMSO-d6):δ164.4(C-2), 103.7(C-3), 182.6(C- 4), 159.96(C-5), 99.1(C- 6), 163.7(C-7), 94.5(C-8), 157.8(C-9), 103.7(C-10), 123.4(C-1′), 128.3(C-2′), 121.4(C-3′), 161.9(C- 4′), 116.6(C-5′), 131.8(C- 6′), 164.3(C-2″),103.4(C-3″), 182.2(C- 4″), 161.0(C-5″), 104.1(C- 6″), 162.7(C-7″), 104.1(C-8″), 155.0(C-9″), 103.4(C-10″), 120.4(C-1‴), 128.4(C-2‴,C- 6‴), 114.9(C-3‴,C-5‴), 161.0(C- 4‴), 55.9(4′-OMe)。综合以上波谱数据,并与文献[26~27]对照,且化合物8与白果黄素标准品共薄层多体系展开,呈现单一斑点,进一步确定该化合物为白果黄素(bilobetin)。
通过对竹柏叶化学成分进行研究,从中分离得到了8个化合物(化学结构见图5),均为黄酮类化合物,其中黄烷类化合物2个(化合物1和3),黄酮碳苷2个(化合物4和5),双黄酮3个(化合物6、7、8)和黄酮类化合物1个(化合物2)。除化合物2、6和7外,其他5个化合物均为首次从竹柏中分离得到,化合物4、5和8为首次从竹柏属植物中分离得到。

图5 分离的竹柏叶化学成分的结构
3 结 论
3.1以吸附率和解吸率为指标,比较了聚酰胺树脂(PLA-1和PLA-2)和大孔吸附树脂(AB-8和D101)对竹柏总黄酮的吸附-解吸性能,结合静态及动态吸附-解吸实验,得出AB-8大孔树脂分离纯化竹柏总黄酮的最佳工艺为:上样液总黄酮质量浓度为1.5~3.0 g/L,上样流速22 mL/h,上样量220 mL;洗脱剂为体积分数80%的乙醇,洗脱流速44 mL/h,洗脱剂用量140 mL,竹柏总黄酮的质量分数从13.6%提高到52.8%,穗花杉双黄酮的质量分数从0.1%提高到2.37%,较纯化前有明显的提高。
3.2从竹柏枝叶提取物正丁醇萃取部位分离得到8个黄酮类化合物,分别为儿茶素(1)、芹菜素(2)、表儿茶素(3)、牡荆苷(4)、牡荆苷鼠李糖苷(5)、穗花杉双黄酮(6)、罗汉松双黄酮A(7)和白果黄素(8)。其中化合物4、5和8为首次从竹柏属植物中分离得到,这为今后对竹柏中黄酮类化合物的定量研究和定性分析提供了参考依据。
