构建诱导型CRISPR/Cas9 系统用于小鼠免疫细胞基因功能研究
2021-04-13赵艳娜唐元家
赵艳娜,邱 荣,沈 南,唐元家
1.上海交通大学医学院附属仁济医院风湿病科,上海市风湿病学研究所,上海200127;2.中国科学院上海营养与健康研究所,上海200031
成簇规律间隔短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR)及其相关蛋白(CRISPR-associated protein,Cas)组成的CRISPR/Cas 系统最早是在古细菌进化过程中形成的RNA 介导的适应性免疫防御机制,能够抵御病毒及外源DNA 的入侵[1]。CRISPR/Cas 系统分为Ⅰ、Ⅱ、Ⅲ型[2-3],Ⅰ型和Ⅲ型系统识别和切割核酸序列需要大型的多Cas蛋白复合物[4],而Ⅱ型系统只需要Cas9 核酸酶和单链导向RNA(single guide RNA,sgRNA)即可。在该系统中,Cas9核酸酶在sgRNA 的指导下对靶基因组DNA 进行切割并引起DNA双链断裂[5-6]。细胞通过非同源末端修复或同源重组修复双链断裂,修复过程中会引起DNA 片段的插入、缺失或替换,从而造成移码框突变,达到基因敲除的目的[7]。
目前,该技术已广泛应用到动物模型建立、哺乳动物发育研究中[8]。特别是近些年Cas9 转基因小鼠的出现解决了Cas9 递送的问题,极大地拓展了CRISPR/Cas9 系统在免疫学研究中的应用[9-10]。但是,大多数CRISPR/Cas9 系统中Cas9 和sgRNA 通常持续表达[11]。这往往会引起基因组DNA 过度切割,从而增加脱靶效应风险[12-13]。此外,一些关键基因敲除可能会导致细胞发育缺陷甚至胚胎致死。因此,为了研究这些基因的功能,往往需要构建条件敲除小鼠或者诱导型基因敲除小鼠,实现特定发育阶段或者特定时间点基因敲除。现在逐渐发展起来的可诱导性基因敲除技术通过诱导性表达Cas9或者sgRNA 来实现基因诱导型敲除,似乎能够有效克服上述局限性[14-15]。
四环素(tetracycline,Tet)诱导型表达系统由于具有高效性、精确性、可控性等优点,广泛应用于基因功能研究,例如基因过表达、基因干扰等[16-17]。其基本原理是通过加入诱导药物Tet 或强力霉素(Dox)改变调控蛋白的构象,从而调控目的基因的表达。在本研究中,通过构建Dox 诱导型sgRNA 表达载体和Cas9 转基因小鼠,建立诱导型CRISPR 系统,可通过体外或体内添加Dox 诱导sgRNA 表达,实现基因诱导性敲除,以期为小鼠免疫细胞内基因功能研究提供一个高效、简单、可控的基因编辑工具。
1 对象与方法
1.1 实验动物、主要试剂及仪器
1.1.1 实验动物 健康、SPF级Rosa26-LSL-Cas9(024857)小鼠和B6.FVB-Tg(EIIa-cre)C5379Lmgd/J(003724)小鼠20只,均购于美国The Jackson Laboratory。本研究实验动物选用6~8周龄雄性小鼠,体质量20~25 g。实验动物均饲养在中国科学院上海生命科学研究院SPF级动物房[动物使用许可证号为SYXK(沪)2019-0001],饲养温度22~25 ℃,空气相对湿度40%~60%。相关动物实验均获得中国科学院上海生命科学研究院医学研究伦理委员会批准(审批号201903H486)。
1.1.2 主要试剂和仪器 限制性内切酶Bbs1、同源重组酶NEBuider HiFi DNA Assembly Master Mix 和T7 核酸内切酶Ⅰ(T7 endonuclease Ⅰ,T7EⅠ)(NEB,美国),Lipo2000(ThermoFisher,美国),小鼠骨髓谱系发育阴性细胞分离试剂盒(美天旎,德国),抗小鼠F4/80 PE 标记抗体BM8(Biolegend,美国),细胞培养基IMDM、无血清培养基Opti-MEM、胎牛血清(fetal bovine serum,FBS)、胰酶(Gibco,美国),细胞因子促血小板生成素(thrombopoietin,TPO)、干细胞因子1(stem cell factor 1, SCF1)、 白 介 素-6 (interleukin-6, IL-6) 和IL-3(R&D,美国),Plat-E 细胞和L929细胞购自中国科学院细胞库,超净工作台、CO2培养箱(Thermo Scientific,美国),光学显微镜(Nikon,日本),荧光显微镜(ZEISS,德国), Centrifuge 5417R 低温高速离心机(Eppendorf,德国),DNA Thermal Cycler 9700 PCR 仪(Thermo Scientific,美国),CytoFLEX LX 流式细胞仪(Beckman Coulter,美国)。
1.2 方法
1.2.1 诱导型sgRNA 反转录病毒载体的构建 限制性内切 酶Xho1 和Sal1 双 酶 切LMP ( MSCV-LTR-miR30-PIG) 载体,切胶回收大片段获得反转录病毒载体骨架。基因合成U6-TetO-sgRNA 和EF1α-T2A-Puro-BFP-T2ATetR 片段(苏州金唯智生物科技有限公司)。利用同源重组将2个片段组装进反转录病毒载体骨架,经过转化、挑克隆、测序,获得正确载体。
1.2.2 sgRNA 设计 使用MIT CRISPR 设计软件设计sgRNA (http://crisp.mit.edu)。F4/80 sgRNA 序列: 5'-ACCCAAGATCCATTACAATG-3'。
1.2.3 反转录病毒包装和细胞系感染 反转录病毒由质粒转染Plat-E 包装细胞产生,按照Lipo2000 说明书进行转染。转染体系:20 μg 质粒DNA 加入到500 μL Opti-MEM 中稀释DNA(溶液A)混匀;40 μL Lipo2000 加入到500 μL Opti-MEM(溶液B)混匀;将溶液A 和B 混合混匀,室温静置15 min 后加入培养皿。4 h 后换液,48 h收集病毒上清液。反转录感染NIH3T3 细胞系,需提前1 d将细胞接种于96孔板,次日待细胞汇合度达到30%时即可进行病毒感染,感染48 h 后,流式细胞术检测病毒感染效率。
1.2.4 小鼠骨髓造血干细胞的分离 颈椎脱臼法处死小鼠取其骨髓细胞进行后续实验。采用Miltenyi MACS免疫磁珠谱系阴性分选试剂盒,根据说明书的方法富集小鼠骨髓谱系发育阴性细胞(Lineage negative cell,Lin-)。
1.2.5 反转录病毒感染小鼠骨髓造血干细胞 小鼠Lin-细胞培养基为IMDM,含10%FBS 并加入细胞因子(TPO 50 ng/mL、SCF1 50 ng/mL、IL-6 50 ng/mL 和IL-3 10 ng/mL)。细胞激活48 h 后进行反转录病毒感染,收集病毒悬液加入6 μg/mL聚凝胺混匀,然后使用该病毒混合液重悬造血干细胞,1 000×g,32 ℃离心90 min。离心结束后,吸去病毒上清液,补加2 mL 含细胞因子的培养基继续培养。病毒感染48 h 后,流式细胞术检测蓝色荧光蛋白(blue fluorescent protein,BFP)阳性细胞比例。
1.2.6 小鼠骨髓来源的巨噬细胞诱导培养和Dox 诱导sgRNA表达 为了验证诱导型CRISPR系统是否可以实现诱导性基因敲除,选择巨噬细胞的表面分子F4/80 进行实验。分离Cas9转基因小鼠骨髓细胞,用含10%L929上清培养基培养小鼠骨髓细胞,诱导其向巨噬细胞分化。第2日分别感染NC-sgRNA 和F4/80-sgRNA 病毒。第3 日,根据实验设计加入Dox 诱导sgRNA 表达,实验分为NC Dox-、NC Dox+、F4/80 Dox-和F4/80 Dox+共4 组,并加入Puromycin 筛杀未感染的细胞。第7日,收集细胞进行F4/80抗体染色,通过流式细胞术检测F4/80阳性细胞比例。1.2.7 T7EⅠ检测基因组DNA 切割效率 使用细胞抽提基因组DNA 试剂盒提取基因组DNA,利用Pusion聚合酶PCR扩增F4/80 sgRNA靶位点或预测的脱靶位点周围的基因组区域。F4/80 sgRNA-F:5′-AACGCTTGGGTTTTAT TCGTAGC-3′;F4/80 sgRNA-R:5′-CTTAAGGCCGACT ACGTGCTG-3′。PCR 扩增待检测DNA 片段,PCR 产物电泳后切胶回收目的条带。将纯化PCR产物与Buffer 2混合进行梯度退火。结束后加入T7EⅠ,并将混合物在37 ℃下孵育1 h。最后加入0.25 mol/L 乙二胺四乙酸以终止反应,产物在2%琼脂糖凝胶上进行凝胶电泳。
1.2.8 流式细胞仪检测及分析 使用CytoFLEX LX 流式细胞仪检测BFP 和F4/80 表达。使用FlowJo 软件(Becton Dickinson)进行数据分析。
1.3 统计学方法
使用GraphPad Prism 5 软件对实验结果进行统计学分析。2 组独立样本数据比较采用非配对t 检验。P<0.05 表示差异有统计学意义。
2 结果
2.1 构建Dox诱导型sgRNA表达载体
为了建立药物诱导型CRISPR/Cas9 系统,实现基因诱导性敲除,构建Dox 诱导型sgRNA 反转录病毒表达载体。该病毒载体主要由U6-TetO-sgRNA 和EF1α-T2APuro-BFP-T2A-TetR 组成(图1A)。当系统中不存在Dox情况下,Tet 阻遏物(TetR) 结合在Tet 操纵子位点(TetO)上,严格抑制U6启动子活性,阻遏sgRNA 表达。当加入Dox 后,可解除TetR 对TetO 的抑制,快速有效地启动该启动子活性,诱导sgRNA 的表达,Cas9 和sgRNA结合后可进行基因编辑,从而实现基因可诱导性敲除(图1B)。
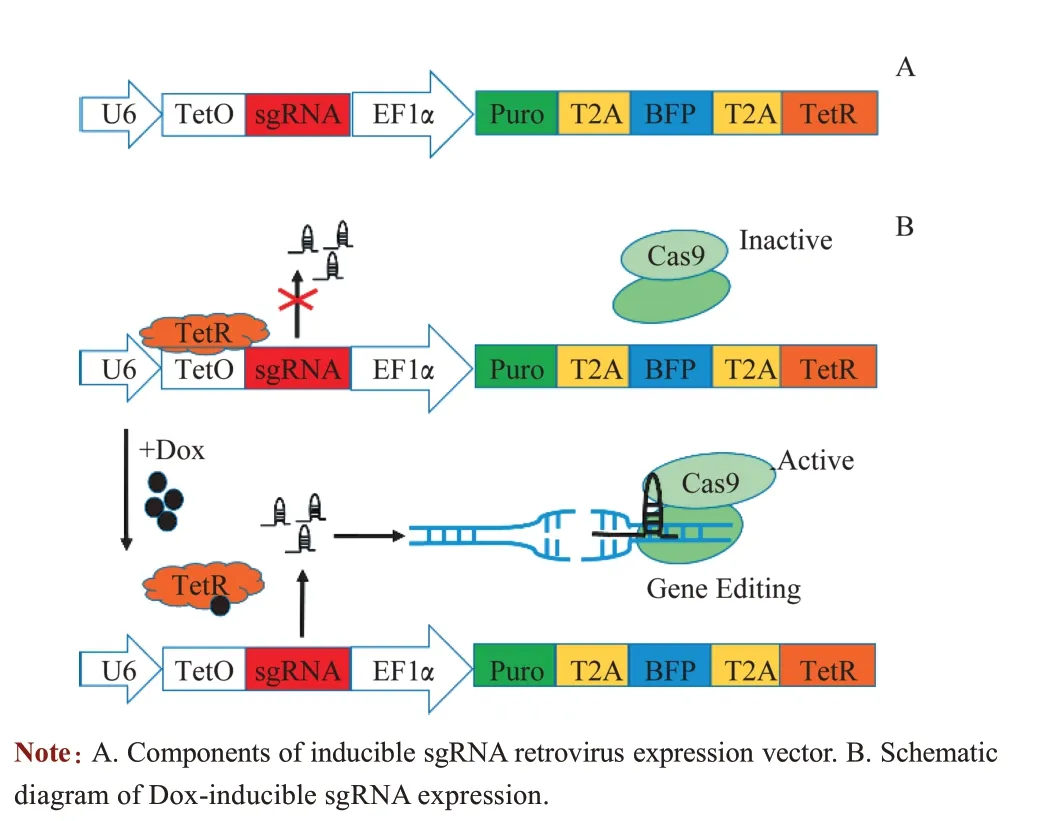
图1 Dox诱导型sgRNA表达载体构建策略Fig 1 Construction strategy of Dox-inducible sgRNA retroviral vector
2.2 反转录病毒包装和NIH3T3 细胞感染
为了验证反转录病毒载体是否构建成功并能够产生具有感染性的病毒颗粒,经测序验证后,进行病毒包装和细胞感染。本研究采用Plat-E细胞进行病毒包装,只需要转染病毒载体质粒即可产生表达特定sgRNA的反转录病毒。转染24 h后,显微镜下观察细胞状态,结果发现细胞形态正常(图2A)。转染48 h后,在荧光显微镜下观察到BFP正常表达(图2B)。为了验证是否可产生具有感染性的病毒颗粒,收集48 h病毒上清液,感染NIH3T3细胞。感染48 h后,收集细胞,流式细胞术检测BFP阳性细胞比例,结果显示BFP 阳性细胞比例高达50%(图2C)。以上结果说明Dox诱导型sgRNA反转录病毒表达载体构建成功,并且可以产生具有感染性的反转录病毒颗粒。
2.3 Dox诱导巨噬细胞中F4/80基因敲除
流式细胞术检测结果显示:在NC Dox-、NC Dox+和F4/80 Dox-组中,几乎没有F4/80 阴性群体;而在F4/80 Dox+组中,F4/80 阴性群体比例高达50%(图3A)。对4组数据进行统计学分析,结果(图3B)显示:F4/80 Dox+组F4/80 阴性细胞比例与其余3 组比较显著增加,差异具有统计学意义(P=0.000)。以上结果显示,诱导型CRISPR/Cas9系统实现了基因诱导性敲除,其发挥作用严格依赖Dox。

图2 反转录病毒包装和NIH3T3细胞感染Fig 2 Retrovirus packaging and NIH3T3 cell infection
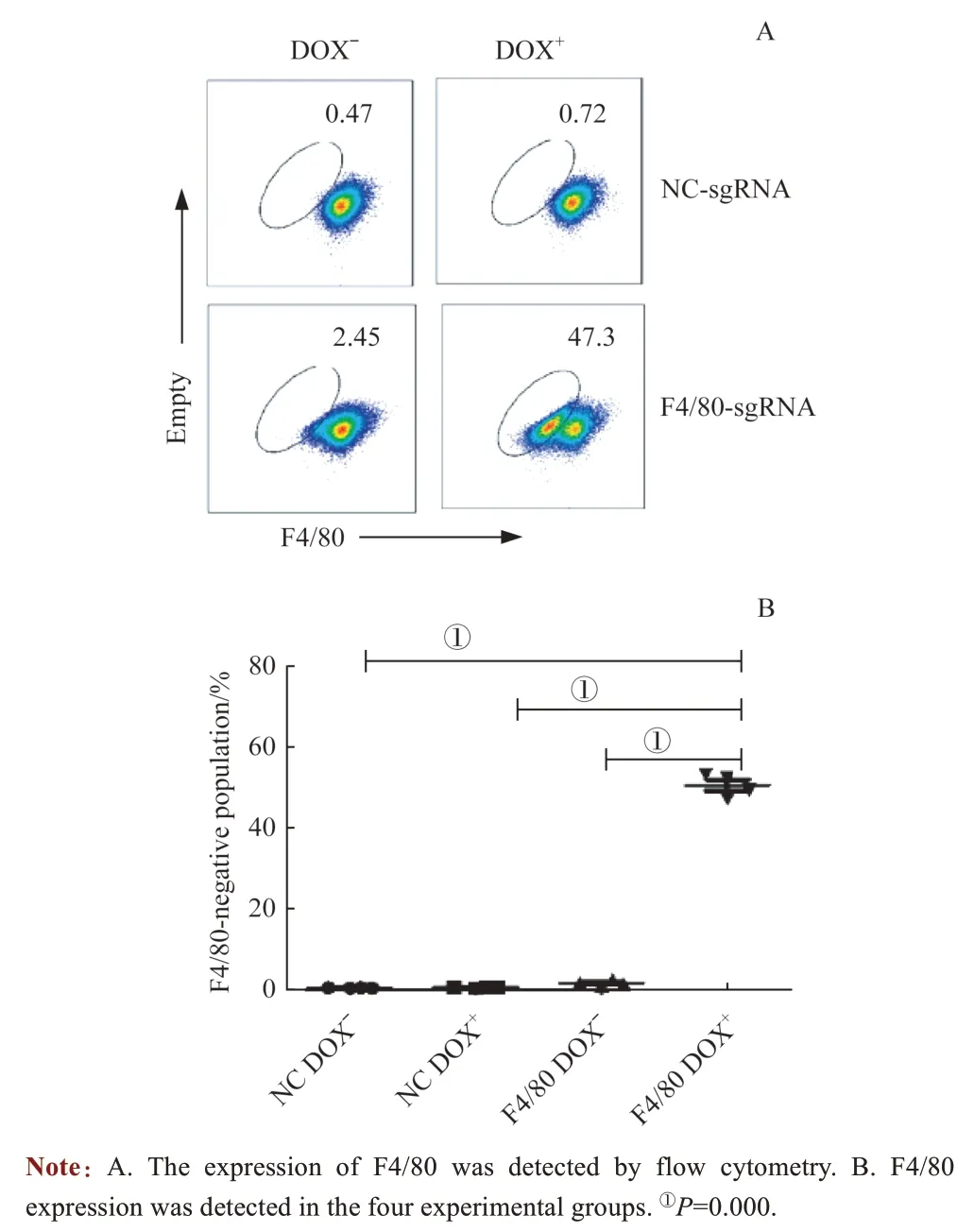
图3 流式细胞术检测骨髓来源的巨噬细胞中F4/80 表达Fig 3 Flow cytometry detection of F4/ 80 expression in bone marrow-derived macrophage
2.4 T7EⅠ实验检测基因组DNA编辑效率
为了验证F4/80基因组DNA是否发生突变,抽提编辑后细胞基因组DNA。围绕F4/80-sgRNA靶向位置上下游设计引物进行PCR扩增,进行后续T7EⅠ实验。T7EⅠ实验结果显示:在F4/80 Dox-组中,T7EⅠ实验产物是完整的,未发生切割;与之相反,在F4/80 Dox+组中,T7EⅠ实验产物被切割为约250 bp和230 bp的2条带,大小符合预期(图4)。以上结果显示,诱导型CRISPR/Cas9 系统实现基因诱导性敲除,其发挥作用严格依赖Dox。

图4 T7EⅠ实验检测F4/80基因组DNA编辑效率Fig 4 Detect DNA mutations of F4/80 locus by T7EⅠ
2.5 反转录病毒感染小鼠骨髓造血干细胞
利用免疫磁珠分选试剂盒分离小鼠骨髓造血干细胞,激活48 h 后,进行反转录病毒感染。流式结果显示,病毒感染初始2 组BFP 阳性细胞比例为25%左右,加入Puromycin 进行药物筛选后,BFP 阳性细胞比例接近100%(图5)。以上结果显示,利用反转录病毒可以成功感染小鼠骨髓造血干细胞。

图5 反转录病毒感染小鼠骨髓造血干细胞Fig 5 Retrovirus infection of mouse hematopoietic stem cells
3 讨论
近些年发展起来的CRISPR/Cas9 系统能够以高效、特异、准确的方式实现基因组编辑。除了利用CRISPR/Cas9技术研究单个基因功能外,CRISPR高通量筛选在发现多种生物学过程表型调节分子中也发挥着重要的作用,例如耐药性、基因表达、代谢和病毒感染的必需因子。但是,利用CRISPR/Cas9 系统进行体内基因功能研究时仍然存在一些局限性。例如,体内进行造血干细胞功能研究时,当基因功能对细胞存活必不可少时,将难以产生可利用的细胞。本研究通过构建Dox 诱导型sgRNA 表达载体结合Cas9 转基因小鼠,建立了诱导型CRISPR/Cas9 系统。在该系统中,可以通过添加Dox 诱导sgRNA表达,从而在Cas9小鼠细胞中成功实现诱导性基因敲除,为小鼠免疫细胞基因功能研究提供了可控的工具。
之前也有大量研究尝试建立一种可诱导型CRISPR/Cas9 系统,如有研究[18]开发了一种可用光激活的CRISPR/Cas9系统;但是,大多数实验室都没有专用的照明设备用于活化Cas9 蛋白。此外,一些研究尝试将Cas9基因置于Tet 响应元件启动子控制下,但后来被证明有Cas9泄漏表达,从而引起大量不受控制的基因敲除[19-20]。另有一项研究开发了一种Dox 诱导型gRNA(gRNAi)AAV 载体[21],该载体包括H1/TetO 启动子和持续表达TetR等结构。此外,该研究发现不同长度的H1/TetO启动子和U6/TetO启动子可以通过Dox依赖方式实现相似基因组编辑效果。该研究成功运用该gRNAi 载体在小鼠脑内神经元中实现可控的基因组DNA 编辑。在这个系统中,只需要向动物喂食1 d 含Dox 食物即可诱导体内发生基因组编辑,证明诱导型CRISPR/Cas9 系统在体内基因功能研究中具有广泛的应用前景。
造血干细胞具有自我更新能力和增殖能力,可以分化成为红细胞、白细胞、血小板等各个类型细胞;作为血液系统中的“始祖细胞”,被广泛应用于血液、免疫和肿瘤等疾病的治疗。因此,开发一种可以高效研究造血干细胞功能的工具至关重要。有研究开发了一种基于CRISPR/Cas9的体内筛选系统[22]。该研究利用慢病毒载体系统感染Cas9转基因小鼠骨髓造血干细胞并移植到免疫缺陷小鼠中,可以在先天免疫细胞和后天免疫细胞中实现基因敲除。但是,该系统可能存在一定缺陷,如果目的基因对于细胞发育存活是必需的,将无法得到特定发育阶段的细胞,更无法研究该基因敲除后对细胞功能的影响。而本研究构建的药物诱导型CRISPR/Cas9系统,可通过Dox依赖方式在特定发育阶段敲除目的基因,或许可以解决这一难题。
综上所述,在本研究中,通过构建Dox诱导型sgRNA表达载体和Cas9 转基因小鼠,建立了诱导型CRISPR/Cas9系统。利用该系统成功在小鼠骨髓诱导的巨噬细胞中实现F4/80 基因可诱导性敲除。此外,本研究利用该载体产生的反转录病毒成功感染小鼠骨髓造血干细胞,为将来在小鼠体内实现基因可诱导性敲除提供了可能。
