硅酸钴锂正极材料的研究现状与前景
2021-04-02武金磊马运昌魏福祥
武金磊,马运昌,魏福祥,张 翼
(中北大学,山西太原038507)
由于能源和环境问题日益突出,基于电动汽车和电网储能的关键技术引起了世界各国的关注,迫切需要开发出具有高能量密度和大功率的新一代锂电池。电动汽车和大型储能装置的工作电压是由多个单节锂电池通过串联和并联的方式连接提供的。目前市场上主流的单节锂电池型号为18650 或21700 型,并且单个锂电池的电压约为3.7 V。因此,开发具有高能量密度的锂电池的关键是开发具有高电压的正极材料,因为只有单个锂电池的工作电压提高,才可以减少大型储能装置中锂电池的消耗,并有效地提高能量密度。
聚氧阴离子橄榄石LiFePO4材料已经成功商业化,具有很高的安全性和库仑效率(实际充放电比容量已接近其理论比容量[1]170 mAh/g),但由于其固有的结构缺陷(每个配方单元只能提供一个Li+插入和脱嵌[2]),限制了其应用。传统的正极材料不能满足高能量密度锂离子电池的市场需求。在这种情况下,同为聚阴离子系的硅酸盐正极材料引起了研究者的很大兴趣,每单元正硅酸盐Li2MSiO4(M = Fe,Co,Mn ...)可以允许两个Li+插入和脱嵌,并且其Si-O 共价键比P-O 共价键更强,因此在电解液中具有较高的化学稳定性[3]。在研究过程中,发现硅酸盐正极材料具有以下缺点:(1)导电性差,小于10-14S/cm,比LiFePO4低3~5 个数量级[4]。(2)Li2FeSiO4在充放电过程中,第二次循环的充电曲线相对于首次循环会显示出明显的电压降。根据充电前后的X 射线衍射(XRD)分析,发现Li2FeSiO4在第一次充电过程中的XRD 特征峰发生偏移并逐渐稳定,表明该材料在充电过程中发生了相变,从结构无序转变为有序,形成更稳定的结构,且Li2MnSiO4材料在充放电过程中,结构会从有序变为无序,无法恢复。(3)可逆容量低,充放电过程中伴随着氧阴离子的氧化还原(O2-/On-),很难实现两个锂离子的插入和脱嵌。与其他正极材料相比,Li2CoSiO4(LCSO)具有电压平台高(高于4 V)、理论比容量高(为325 mAh/g)、电压平台循环稳定和热稳定性良好等优点,有望成为新一代锂离子电池正极材料。反萤石结构的Li6CoO4材料具有四面体对称结构,以过渡金属氧四面体TO4为结构单元,每个化学式有六个锂位,比硅酸盐具有更高的锂位与变价活性元素摩尔比,从而在理论上具有成倍提高比容量的可能性。以四面体MO4基元为出发点探索新型高锂氧化物正极材料的设计不仅具有学术上的研究意义,同时也将为解决锂离子电池的产业瓶颈奠定科学基础。其首次充电比容量可达到600 mAh/g 以上,但它们与硅酸盐正极材料面临着相似的问题,具有电导率低(小于10-14S/cm)、电压平台低、可逆容量低(首次放电比容量只有27 mAh/g)和结构不稳定等缺点。因此,对于LCSO 正极材料的研究对今后开发出具有更高容量的高锂材料Li6CoO4也具有重要意义。
本文聚焦LCSO 的多晶型结构,通过不同的合成方法和手段改善其物理和电化学性能,总结其近年来的研究进展。
1 Li2CoSiO4的合成
1.1 合成方法
(1)固相反应。传统的固相反应是合成Li2MSiO4/C 最常用的方法,该方法具有成本低、效率高、可延展性好等优点。在固相反应过程中,将反应物充分混合、研磨得到前驱体,然后加热得到Li2MSiO4/C 样品。常用的起始合成材料是含有锂、铁、锰、钴和硅酸盐元素的化合物,例如氧化物、水合物、硅酸盐/硫酸盐/氯化物、草酸盐、乙酸酯、碳酸盐等。有时也使用正硅酸四乙酯[TEOS、Si(OC2H5)4]和Li2SiO3。可添加炭黑、蔗糖、葡萄糖、抗坏血酸、煤、沥青[5]、碳纳米球(CNS)、碳纳米管(CNT)[6]、还原氧化石墨烯(RGO)、氧化石墨烯(GO)、乙炔黑[7]等碳源,以提高正极材料的导电性。采用这种技术,Li2MSiO4通常在600~900 ℃高温下煅烧8~12 h 下合成,但除研磨和退火工艺外,固相反应存在组分混合不均匀、形貌不规则、颗粒生长不受控制、团聚严重、加热时间长等缺点。
(2)溶胶-凝胶法。溶胶-凝胶法以无机材料或金属醇盐为前驱体,在液相中均匀混合,进行水解和缩合化学反应。柠檬酸、蔗糖、聚乙二醇、聚乙烯醇、酒石酸和抗坏血酸等螯合/络合剂常用于溶胶-凝胶技术。合成温度通常为600~800 ℃,反应时间为5~15 h,通过溶胶-凝胶技术可以得到不同形貌的Li2MSiO4,如球形、介孔、团聚体、蜂窝状等。但是用该方法合成的凝胶结构在热处理过程中易崩塌,其原因是加热速率过快和Li2MSiO4结晶度低[8],导致其在高倍率充放电下循环稳定性差。为了克服这些问题,水热合成受到了研究者的高度重视。
(3)水热反应。目前制备Li2CoSiO4的主要方法是水热法合成。水热反应具有简单、清洁、成本低、能耗低等优点,适用于在密闭条件下制备高质量的多晶材料系统[9]。一般来说,水热过程在150~200 ℃的溶剂中进行24~96 h。Gong等[10]首次采用水热反应合成Li2CoSiO4,并以去离子水为反应介质溶剂,获得的Li2CoSiO4由约2 μm 的单个颗粒组成,初始放电比容量为75 mAh/g。Lyness 等[11]提出了以去离子水和乙二醇为溶剂的水热合成锂硅源的新方法,同时对合成材料的结构给出了明确定义,所获得的材料被定义为βⅠ-Li2CoSiO4(Pbn21)可提供60 mAh/g 的初始放电比容量。He 等[12]报道了以碳/二氧化硅(MCS)为模板制备介孔Li2CoSiO4复合材料,但是它的放电比容量只有33 mAh/g。
张志峰等[13]总结了不同的水热方法并进行了改进。通过改进的水热反应合成纯度较高的βII-Li2CoSiO4多晶型材料,使用LiOH·H2O、SiO2和CoCl2·6 H2O(4∶1∶1)作为合成初始材料。在该合成中,将去离子水用作锂和硅源的溶剂,将乙二醇用作钴源的溶剂,去离子水与乙二醇的比例为2∶1,然后将两种溶液混合并搅拌以形成Li2CoSiO4前驱体,再将前驱体转移至高压反应釜中,在150 ℃下加热72 h,得到的材料由50 nm 左右的单个颗粒组成。与传统的水热法相比,该技术合成的材料具有较小的粒径且分布均匀。杜文强等[14]在张志峰水热反应的基础上,通过借鉴Xu 等人对LiMn0.6Fe0.4PO4的研究[15],发现通过改变水热反应中LiOH 和SiO2的浓度以及乙二醇(EG)与去离子水的比例,能够合成出具有不同形貌的LCSO 材料。合成路径如图1 所示。通过改变去离子水与乙二醇的比例可以改变水热反应过程中混合物的粘度。在较高的粘度下,纳米颗粒倾向于长成次级颗粒的大聚集体,低粘度促进分散的初级纳米颗粒易于形成小尺寸的纳米结构,因此通过改变反应物的黏度可以合成出具有不同形态的LCSO颗粒。通过控制前驱体溶液的[OH-]浓度和黏度,成功合成出了点状、条状和片状的LCSO/C 纳米颗粒。

图1 两步法合成Li2CoSiO4/C 纳米复合材料示意图
(4)其他合成方法。微波热合成法[16]可以让目标材料在短时间内直接吸收电磁/微波能量(自加热),这种方法与其他方法相比耗能较低,具有简便且高效的优点,可以在600~700 °C 下反应6~20 h 制备出纳米团聚体或球体材料,在室温以上(60°C)可获得具有高离子传导性的正极材料。彭忠东等在Li2FeSiO4合成中引入微波外场的作用,通过高能球磨结合微波加热制备Li2FeSiO4/C 材料。其他合成方法还包括原位模板法[17]、喷雾热解法、超临界流体法[7]等。
1.2 碳包覆
导电碳涂层是提高Li2CoSiO4电子导电性的有效而简单的策略。此外,碳包覆还可以防止颗粒接触并抑制颗粒聚集。通常,碳包覆方法有两种。一种是原位碳包覆:混合前驱体和碳源,然后在高温下煅烧,该方法可防止颗粒团聚,获得具有均匀碳涂层的复合材料。另一种是非原位碳包覆:首先合成不含碳的原料,然后与碳混合。原料与碳源通过球磨/研磨混合。如果碳源不是炭黑,则需要进行热处理。目前常见的碳源也分为两类,一类是有机碳源,如柠檬酸、纤维素、蔗糖和聚乙二醇(PEG),另一种是无机碳源,例如炭黑。
早期,研究者使用炭黑和石墨作为Li2CoSiO4正极材料的碳源,并通过球磨方式将其混合。然而,仅通过球磨方式混和碳源和Li2CoSiO4前驱体获得的Li2CoSiO4/C 复合材料的电化学性能并没有明显改善。之后人们陆续尝试了几种不同的碳涂覆方法,包括在2013 年使用有序的介孔碳/二氧化硅(MCS)框架作为硅酸盐前体和碳源以及在2017 年使用多壁碳纳米管[18],以改善Li2CoSiO4的电化学性能,但都效果不大。
直到2018 年,张志峰等人以蔗糖为碳源,通过球磨的方式将蔗糖与Li2CoSiO4材料混合,Li2CoSiO4通过水热反应制备,将Li2CoSiO4与蔗糖的混合物送入管式炉在600 ℃下煅烧1 h。合成方法如图1 所示,通过这种碳包覆方法获得的Li2CoSiO4/C 复合材料的电化学性能得到较大提升,首次充放电比容量分别为226 和112 mAh/g,而未进行碳包覆的Li2CoSiO4材料首次充放电比容量仅为82.4 和30.2 mAh/g,电压平台介于2.5~4.6 V[13]。其电化学性能提升的原因在于在高温下由蔗糖碳化形成的碳涂层,不仅在活性颗粒之间提供了导电连接,从而促进了电子和离子的迁移,而且在热处理过程中减小了活性材料的粒径。它可以缩短锂离子的扩散路径,促进相邻粒子之间的良好接触并减少极化。
1.3 掺杂
包覆只能从外部提高硅酸盐材料的导电性,而离子掺杂则是从根本上改变锂离子的内部结构,从而提高锂离子在材料中的扩散效率。从结构稳定性、可逆性、倍率性能和循环等方面研究发现,离子掺杂是提升其电化学性能的有效途径。因此,为了提高Li2CoSiO4的性能,Co 位、Li 位、Si 位和O位等各种掺杂技术在Li2CoSiO4的掺杂中得到了广泛的应用。
硅酸盐正极材料发展缓慢的主要原因是电子和离子导电性差。一些研究人员已经计算出钠化合物具有更高的电子和离子导电性,而锂化合物具有更高的脱锂电压和更好的循环稳定性。因此,为了提高LCSO 的电化学性能,可以在Li位进行适当的Na 掺杂。
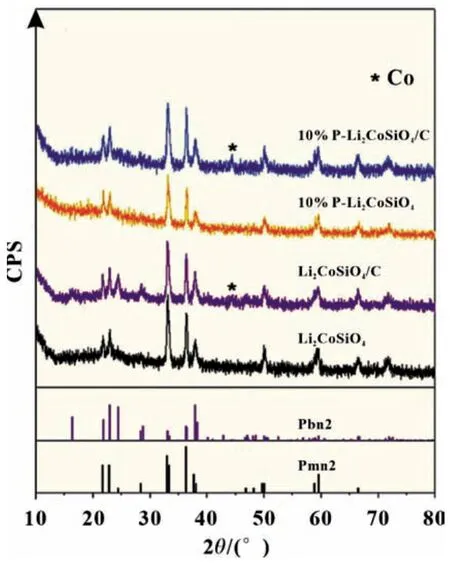
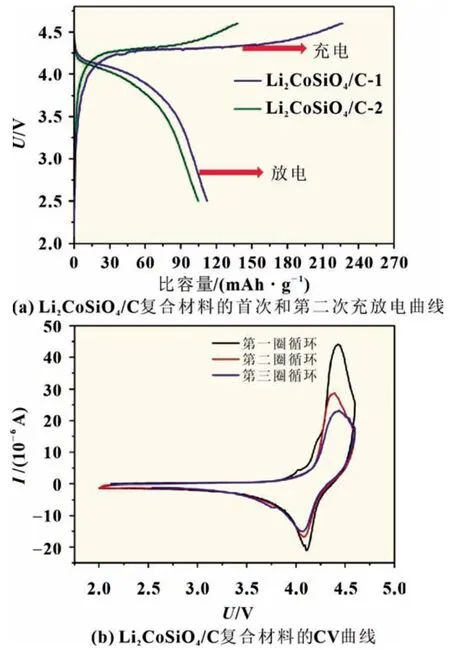
在Co 位进行适当的离子掺杂可以改变Li-O-Co 键的强度,引起材料晶胞体积的变化,但不会改变LCSO 的晶体结构。此外,Co 位掺杂可以降低电化学极化,有利于Li+的插入和提取。Co 位掺杂常用的元素有Fe、Mn 和Mg 等。这个类型可表示为Li2Co1-xMxSiO4(M=Fe,Mn,Mg)(0 Si位掺杂可以抑制SiO44-的强绝缘性,提高LCSO 的电导率,从而提升其电化学性能。张等以磷酸为磷源,通过水热法成功合成了Li2CoSi0.9P0.1O4/C 复合材料[13],其初始充电比容量为270 mAh/g,放电比容量为144 mAh/g。在进一步了解聚阴离子强绝缘性的基础上,杜等[21]通过系统地改变Al 的掺杂量,研究了碳包覆和Al 掺杂对LCSO 电化学性能的影响,发现Li2CoSi0.96Al0.04O4具有最佳的电化学性能,其首次充放电比容量分别为331 和140 mAh/g。表1 对近年来文献中Li2Co-SiO4材料的性能做了汇总。 表1 文献报道的Li2CoSiO4材料第一次充放电比容量 Li2MSiO4化合物具有与Li3PO4相似的结构,都属于四面体构型。它由正交的O2-离子(扭曲的六边形紧密堆积形式)组成,其中阳离子占据四面体空隙的一半。阴离子通过与O2-共顶角连接。这种以四面体氧化物为结构单元的化合物具有丰富的晶体结构。当前存在五种已知类型,可以将其大致分为β 和γ,分别对应于Li3PO4的β 相和γ 相。在β 型结构中,所有四面体指向相同并垂直于O2-的紧密堆积面,并且四面体通过共同的顶部相连;在γ 型结构中,每个四面体都作为结构单元堆叠在一起,中间的四面体的方向与另外两个相反,它们以共线方式连接。根据阳离子在四面体中排列顺序的不同,会有不同的结构和晶型,进而将β 和γ 结构分为βⅠ、βⅡ、γ0、γⅡ和γs。 目前我们知道LCSO 存在多种形态,包括βI、βII和γ0三种不同温度下合成的形态[22],三个相的结构如图2 所示。通过水热法合成得到βII相,然后把βII相在700 ℃高温处理2 h 得到βI相。将βII相1 100 ℃处理2 h,然后降至850 ℃,最后淬火至室温得到γ0相[11]。它们在高温时很容易发生相转换,例如在600 ℃碳包覆时会出现βII相向βI相的转换,导致很难得到βII纯相。LCSO 化合物属于四面体结构。一般在材料的晶体结构中,氧原子呈现出轻微扭曲的密排六边形结构。Li、Co、Si 和氧原子分别形成四面体,其中一半四面体中心被阳离子占据,从而避免四面体之间的面共享。Armstrong 等通过rietveld 精修LCSO 三种不同结构的X 射线和中子粉末衍射图[22],发现βII相中存在明显的阳离子占位无序。在βII相的理想构型中,Co 和Li 分别分布在四面体的2 a 和4 b 处。而实际合成的βII相过渡金属2 a 位置完全被Li 占据,而4 b 的位置基本上被Co 和Li 等同占据,βII相中的Li/Co 混排程度比较剧烈,βI相中的Li/Co 混排程度相对较弱,而γ0相中没有Li/Co 混排。之后通过Li-MAS-NMR 谱(核磁共振谱)进一步证实了这一结果。 图2 Li2CoSiO4 三种不同相的晶体结构 Noriaki 等[23]通过900 ℃高温固相反应合成了具有Pbn21(βI)相的LCSO。从DTA 曲线发现,LCSO 在540 ℃开始由βII相转变为βI相。同时,通过高温原位XRD 证实了Pbn21到Pmn21的可逆二阶相变,解释为Co 和Li 原子的有序无序转变。Li/Co 混排引起Pbn21相有序到无序的转变。Co 亚晶格的无序是由于与Li 原子的位置交换造成的,这与水热反应合成材料的报道一致。过渡金属的有序度直接影响到LCSO 不同相的结构稳定性、高压性能和热处理工艺。Wu 等[24]通过第一原理计算研究了LixCoSiO4(0≤x≤2)的结构和电子性能。结合不同脱锂状态下的形变电荷密度和电子态密度,发现Co-O键的键长随着锂离子的脱嵌和钴离子的氧化显著减小,而Si-O 键的键长基本没有变化。这表明在锂化-脱锂过程中,硅与氧原子的成键性能没有发生变化;同时证明了Li2CoSiO4的结构稳定性实际上是强Si-O 共价键作用的结果。根据Zhang等[25]的计算结果,Co 基硅酸盐的晶格框架不同于铁基和锰基体系的二维层状结构,是一种三维框架结构。脱锂过程中产生的Peierls 畸变可能是制约钴基材料充放电能力提高的关键微观因素。 在第一性原理密度泛函理论(DFT)计算的基础上,研究不同离子掺杂对LCSO 结构的影响,用于指导实验合成。所有结构基于DFT 理论的VASP 软件计算,并采用PAW 赝势,交换关联函数GGA 和PBE 方法。 通过构建P 在LCSO 的Si 位掺杂模型进行研究,LCSO 中P 取代Si 的原子模型用2×1×1 的上层结构构成。该结构包含8 个分子式,其中一个被P 取代。由于P 比Si 少一个电子,因此计算了带负电态的P 掺杂体系。然后,对相关模型进行了第一原理结构优化。通过比较Li1/2CoSiO4和Li1/2CoSi7/8P1/8O4在75%脱锂相中的模型以及CoO4四面体在两种结构中的键长分布可以看出,在未掺杂模型中,Li+具有对称的阶梯分布,四面体形成有序的电荷层,这表明它具有一个带有Peierls 畸变的高应力网络,在P 掺杂模型中,P 掺杂位点可以形成排斥性正电荷中心,从而可以抑制脱锂相Peierls 畸变的非对称应力。同时,通过比较P 掺杂于未掺杂计算模型的电子结构计算,发现P 掺杂可以减小LCSO 的能带间隙[13]。 通过湿式球磨混合LCSO 前驱体和蔗糖并在氩气中于600 ℃煅烧1 h,制备0.1P-LCSO/C,0.04Al-LCSO/C 复合材料,它们表现出主要的正交相(Pmn21-DP)。其XRD 特性如图3 所示(LCSO 在20°~25°之间的XRD 谱中有两个特征峰,在本工作中标记为DP 相,在20°~25°之间的XRD 谱中有三个特征峰被标记为TP 相)。从之前的工作中我们了解到,LCSO 在540 ℃开始从DP 相转变为TP 相,即在未掺杂的情况下,LCSO 在高温碳包覆后表现为DP 和TP 的混合多晶型相。但是我们发现,在LCSO 的Si 位进行P、Al 掺杂后,即使经过600 ℃高温碳包覆后,得到的产物DP 相仍然占主导地位。这表明在Si位进行离子掺杂可以有效地抑制TP 相的产生。以前我们认为Si 位掺杂的主要作用是改变LCSO 的电子传输特性,然而,通过实验发现Si 位掺杂不仅可以改变四面体多晶型的热稳定性,而且有助于获得LCSO 纯相材料。此外,掺杂[13]过程也给我们带来了一些困惑。与纯相材料相比,掺杂后形成的复合材料在碳包覆过程中更容易产生Co 或Co 相关杂质。通过分析,推测TP 相的结构稳定性优于DP 相,而在Si 位进行掺杂后会抑制TP 相的生成,因而导致在Si位进行掺杂后的复合材料相对于未掺杂的LCSO 在碳包覆过程中更易于还原Co。 图3 LCSO、LCSO/C、0.1P-LCSO和0.1P-LCSO/C 的XRD 图谱 图4 Li2CoSiO4/C 复合材料的充放电曲线和CV曲线 在2.5~4.6 V 电压平台内,取电流密度5 mA/g 对LCSO/C复合材料进行充放电测试。Li2CoSiO4/C 复合材料的第一次充电比容量为226 mAh/g,放电比容量为112 mAh/g,具有约4.25 V 的充电平台和约4.1 V 的放电平台,如图4(a)所示。在第二圈循环中,电压平台没有发现明显的变化。与已有的文献报告相比,在2.5~4.6 V 的工作电压内,0.7Li+的可逆脱嵌远高于文献报道的75 mAh/g(0.46Li+)LCSO 的最大容量。 通过对LCSO/C 复合材料的循环伏安(CV)曲线进行分析[26],如图4(b)所示,发现在第一圈循环中LCSO/C 的氧化峰在4.3 V 左右,还原峰在4.15 V 左右;4.3 V 的氧化峰与4.15 V的还原峰代表了LCSO 材料中Co2+离子的氧化和还原,在接下来的两次循环中也存在同样位置的氧化还原峰。而氧化峰和还原峰的强度代表着氧化还原反应的难易程度,峰的强度越大,氧化还原反应越容易。氧化峰和还原峰的强度比值越接近于1,代表电池可逆性越好,放电容量越接近充电容量。从CV 曲线中,我们发现LCSO/C 复合材料的氧化峰比还原峰强得多,代表这个材料的可逆性不是很好,即放电容量远小于充电容量,这个结论可以通过充放电曲线得到证实。同时还发现,在第二圈循环过程中的氧化峰与第一圈循环相比强度迅速下降,这表明材料的循环稳定性较差。体现在在充放电曲线中为第二圈的充电容量远小于第一圈。 对比掺杂P 后材料的充放电曲线和CV 曲线,如图5 所示[26],发现其存在和未掺杂材料类似的情况,即在充放电过程中充电容量远大于放电容量,表现在CV 曲线中为氧化峰强度远大于还原峰,且在随后的循环过程中,氧化峰和还原峰都有不同程度的下降,这应该就是LCSO 材料循环稳定性差的原因。0.1P-LCSO/C 材料的充放电比容量[13]分别为270 和143 mAh/g,基本上能够实现一个锂离子的插入和脱嵌。0.04Al-LCSO/C 的首次充放电比容量[21]分别为331 和140 mAh/g。这表明在Si 位进行P、Al 掺杂在一定程度上虽然能够提升LCSO 材料的电化学性能,但对其循环稳定性改善程度有限。 图5 P-Li2CoSiO4/C复合材料的充放电曲线和CV 曲线 为了搞清楚对Si 位进行掺杂相对于纯相材料电化学性能提升的原因,以及纯相材料电化学性能差的原因,通过XRD 测试了充放电前后的0.04Al-LCSO/C 和经过一次循环的LCSO/C 复合材料,如图6 所示[27]。发现在充放电循环过程中LCSO/C 复合材料的一些XRD 特征峰消失,表明LCSO/C 复合材料的结构在充电过程中发生了有序到无序的转变。同时,由于LCSO/C 复合材料的结构无序化,导致锂离子迁移路径紊乱,难以将锂离子重新插入到LCSO/C 颗粒中,因而它在首圈循环中具有较大的不可逆容量。而且,与未经过充放电测试的原始LCSO/C 材料相比,发现在第一次充放电循环后,LCSO/C 材料的XRD 特征峰强度变弱,表明其结晶度在充放电循环过程中降低。结合充放电曲线分析,LCSO/C 复合材料在放电过程中不能保持长程有序,导致材料的库仑效率低,即放电容量远小于充电容量。另外,由于结晶度连续降低,导致Li2CoSiO4/C 的循环稳定性差。与未掺杂材料相比,掺杂后的材料在充电的末态仍能保持原来的结构,但与未经过充放电测试的材料进行对比,发现它们的XRD 特征峰强度降低,即结晶度比未充电时有所降低。在LCSO 的Si 位进行P 和Al掺杂,与未掺杂材料相比,在一定程度上能够保持结构的稳定性,降低材料在充放电过程中结晶度下降的趋势,从而具有相对较高的充放电性能和循环稳定性。 图6 0.04Al-LCSO/C 和LCSO的XRD 对比图 LCSO 正极材料电化学性能差的根本原因在于其在充放电过程中,结构上会发生有序到无序的转变,虽然这个过程是可逆的,但在充放电过程中,随着循环的进行,其结晶度会不断下降直至消失,导致其循环稳定性差。目前改善LCSO材料电化学性能最有效的措施是在Si 位进行离子掺杂,一方面可以改善SiO44-的强绝缘性,另一方面,能够在一定程度上抑制LCSO 在充放电过程中结构有序到无序的相转变,降低其结晶度下降趋势。但这仍未从根本上解决LCSO 在长循环过程中结构有序到无序的相转变,因而只能在一定程度上改善LCSO 的循环稳定性。今后我们的工作重点是,找到更好的掺杂元素来稳定LCSO 材料的结构。之前我们了解到Li2MnSiO4材料在充放电过程中结构会发生有序到无序的相转变,且不能恢复,但是有研究人员对Li2MnSiO4材料进行Fe掺杂,可以改善其结构稳定性,在充放电过程中保持其结构有序性。这证明,找到合适的掺杂元素,保证LCSO 材料在充放电过程中保持其结构长程有序性是可行的。此外,LCSO材料的低温相存在很大程度的Li/Co 混排,即Li、Co 占位无序,在充放电过程中会影响锂离子迁移路径,进而影响其电化学性能。而通过计算和实验表明,在高温下可以获得占位有序的、具有晶体结构的LCSO 材料,目前尚未发现这方面的研究。这为我们在高温下合成出占位有序的晶体结构材料,从而改善LCSO 的电化学能提供了一定的指导意义。 随着锂离子电池在电动汽车(EVs)和大型电能储存设备上的大规模应用,迫切需要开发出能量和功率密度更高、循环稳定性更好的新一代锂离子正极材料。通过改善合成工艺,使用碳包覆和离子掺杂等改性手段对LCSO 材料的电化学性能进行改善,有望使其成为下一代动力锂离子电池的重要候选材料之一。
2 聚阴离子结构
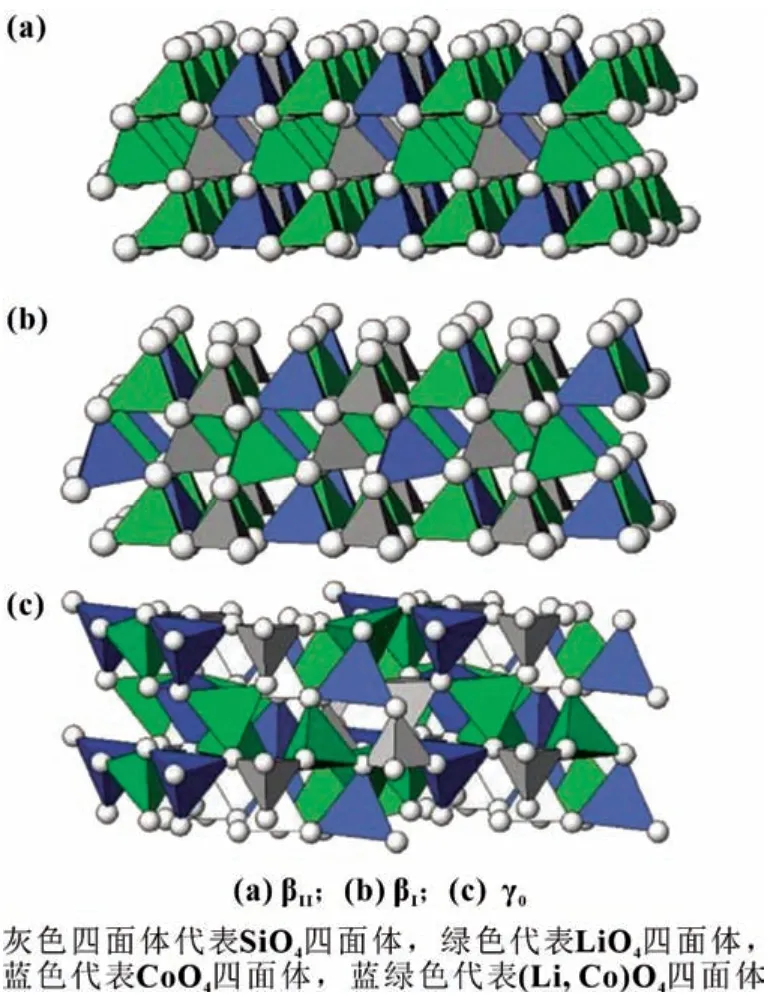
2.1 Li2CoSiO4的多晶结构及其相变
2.2 掺杂对Li2CoSiO4结构的影响
3 电化学性能


4 结论
