未分化甲状腺癌分子机制和治疗进展
2021-03-24陈宝定赵双双陈延玮王珂珂张政袁国跃
陈宝定,赵双双,陈延玮,王珂珂,张政,袁国跃
(江苏大学附属医院 1.超声医学科;2.内分泌科,江苏 镇江 212001)
未分化甲状腺癌(anaplastic thyroid carcinoma,ATC)是一种少见的高度恶性肿瘤,与分化型甲状腺癌(包括乳头状甲状腺癌和滤泡状甲状腺癌)相比,ATC的癌细胞没有保留甲状腺滤泡的生物学功能,例如碘的摄取、甲状腺球蛋白合成和促甲状腺激素的依赖[1]。快速增长的颈部肿块、极高的远处转移率和短暂的生存期是ATC的突出临床特征。迄今为止,对于ATC尚没有标准的治疗方案,常规治疗以外科切除和放疗的综合治疗为主,辅以化疗、免疫、分子靶向及姑息治疗等[2]。ATC起病突然、早期诊断困难、进展迅速,多数病例发现时就已经存在广泛的周围侵犯和远处转移,其中位生存期低于6个月,1年生存率仅为10%~20%,2年生存率为12%左右,5年生存率低于5%[3-4]。
ATC具有多样性和多变性的复杂分子机制特征,目前已发现的与ATC相关突变基因包括TP53、BRAF、RAS、ALK、PIK3CA、CTNMB1和PTEN等主要驱动基因以及几十种相关癌基因,而且这些基因在肿瘤的发展中会不断变化[1]。靶向治疗和免疫治疗是ATC近年研究热点,2018年,美国FDA批准达拉菲尼(Dabrafenib)和曲美替尼(Trametinib)联合应用于ATC并伴有BRAFV600E突变的患者,治疗的部分缓解率和完全缓解率分别为57%和4%[5];2020年Capdevila等[6]首次报道了PD-1抑制剂治疗ATC的Ⅱ期临床试验。基于基因组学和蛋白质组学肿瘤分子机制的深入研究,揭示ATC的更多潜在治疗靶点,有望为ATC提供精准的个体化治疗策略。
1 流行病学、临床特征和诊断
在甲状腺癌中,ATC的发病率仅占1%~3%,但其死亡病例在所有甲状腺癌死亡病例中占比高达14%~39%[7-9]。ATC发病的峰值年龄在60~70岁,大多数病例发病年龄大于50岁,女性较男性高发(6 ∶4)。在乳头状甲状腺癌(papillary thyroid carcinoma, PTC)持续增长的发病率背景下,ATC在所有甲状腺癌中占比呈下降趋势[9-10]。ATC发病呈散发特征,在地方性甲状腺肿地区ATC相对高发[10]。有研究显示,约80%ATC发病于长期罹患结节性甲状腺肿的人群[11]。有部分ATC病例发病于分化型甲状腺癌患者,具体占比分歧较大,从7%到89%[2,12-15]。
美国癌症联合委员会(AJCC)第八版肿瘤分期系统和国际抗癌联盟(UICC)TNM肿瘤分期系统将所有ATC诊断为Ⅳ期[16-17],其中:ⅣA期指甲状腺内的ATC,约占10%;ⅣB期为甲状腺外侵犯或颈部淋巴结转移,占40%~45%;ⅣC期为发生肺、纵隔、骨等远处转移,约占50%[18]。
组织病理学结合免疫组化是ATC常用的确诊手段,细胞病理学不能作为ATC的确诊方法。ATC组织学特点取决于梭形细胞、鳞状或上皮样细胞、巨细胞三种主要细胞成分的构成,表现为以梭形和巨细胞为主的肉瘤样形态,以上皮样细胞为主的癌样形态,或两者混合。超声是ATC最常用的影像诊断方法,表现为实质性肿块、极低回声、不规则的边缘、内部钙化、纵横比>1和颈部淋巴结转移等特征(图1);CT、MRI以及PET/CT等对评估肿瘤远处转移、淋巴结转移及周围侵犯具有重要价值[19]。临床上,ATC需要与甲状腺淋巴瘤、甲状腺肉瘤、低分化甲状腺癌、鳞状上皮甲状腺癌以及髓样癌等肿瘤鉴别诊断[20-21]。
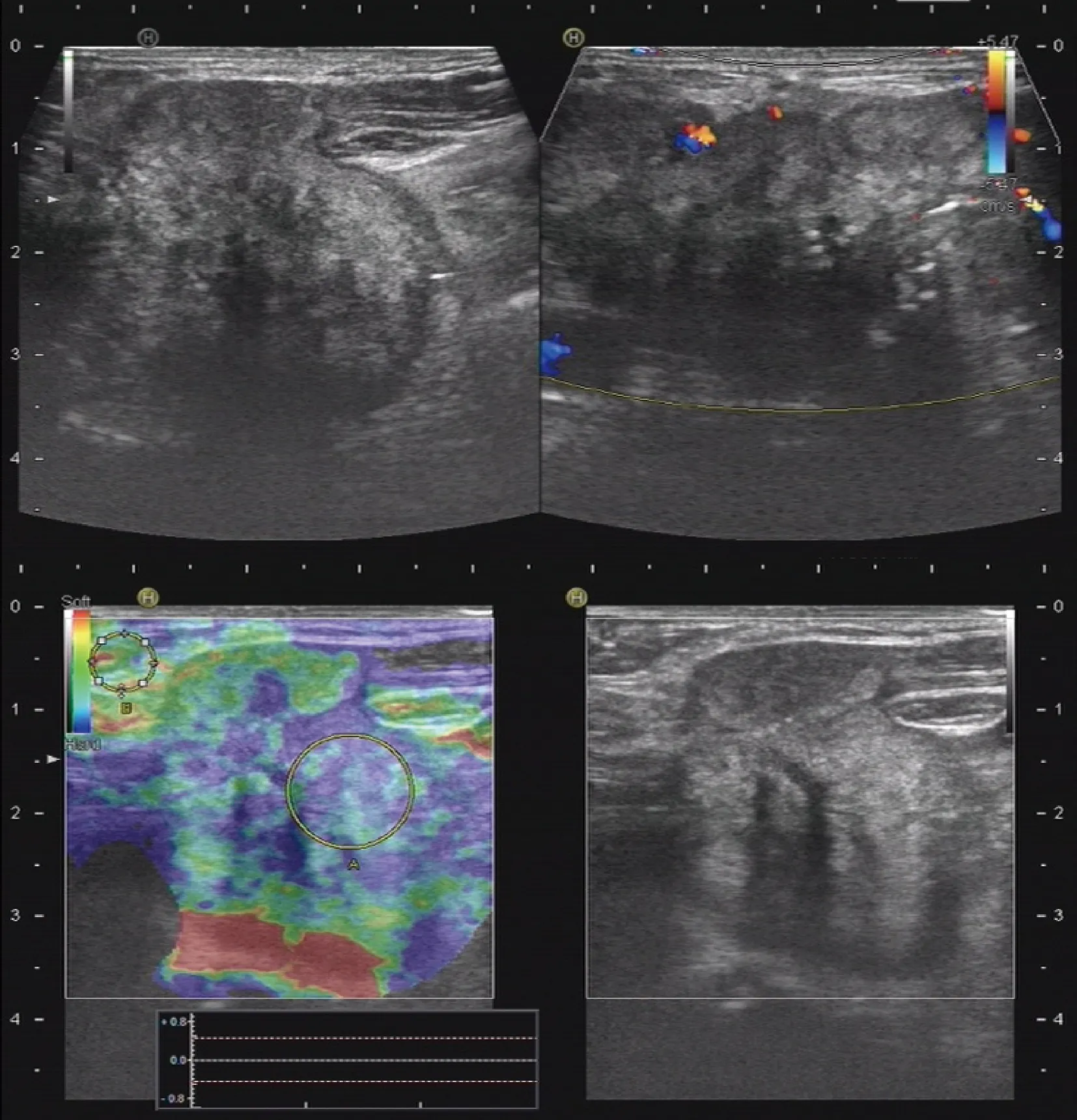
图1 未分化甲状腺癌的灰阶、彩色及弹性超声影像
2 常规治疗方法
ATC常规的治疗方法主要采取手术切除结合体外放射治疗,然后是化疗和姑息治疗的组合[22]。
减瘤切除是ATC常用的外科治疗方法,多数ATC病例发现时就已经广泛周围侵犯和远处转移,根治性手术切除的机会较少,减瘤切除主要用于减少气管的压迫梗阻。对于局限性疾病,如果肿瘤仅限于甲状腺实质,建议在可能情况下进行完整的手术切除。完整的手术切除具有较高的难度,影响因素包括肿瘤的大小、器官外生长以及局部浸润,手术的目标是R0切除(切缘无肉眼或显微镜下肿瘤残留)。术前必须进行高质量的精细断面成像和超声评估,以确定肿瘤的范围以及颈动脉、颈静脉、迷走神经及其分支、气管、食道、胸锁乳突肌和肩带肌群等可能的局部受累,完整的手术切除患者常常有着良好的预后[12]。
由于ATC未分化的表型和缺乏钠碘同向转运体,放射性碘(I131)疗法无法定位腺体,因此只能采用体外放疗,高强度体外放疗对提高生存率的贡献取决于肿瘤是否能够充分切除。一项大样本回顾性研究显示,相较于单纯手术患者,联合术后放疗显著提高了患者的生存率。此外,研究还发现ⅣA期和ⅣB期患者可能在接受术后放疗后延长生存时间,而ⅣC期患者难以改善生存时间[23]。
针对转移性ATC,紫杉烷类(紫杉醇或多西紫杉醇)、蒽环类药物(多柔比星)和铂(顺铂或卡铂)被认为是最有效的化疗药物[12]。对于ⅣA/ⅣB期ATC,如果可能应进行全甲状腺切除术结合体外放疗,进一步推荐的化疗方案包括紫杉醇和卡铂组合、多西紫杉醇和多柔比星组合、单独用紫杉醇或多柔比星[8]。研究显示,联合化疗的效果要优于单独用紫杉醇或多柔比星[1]。
由于ATC极易累犯周围组织及发生远处转移,总体预后仍然较差。多项研究表明,常规治疗方法并不能有效阻止ATC的疾病进程[1-2,13,24]。
3 分子机制
对于驱动ATC发病的分子机制和指导个体化精准治疗靶点/多靶点的研究是目前研究的热点, 新的分子机制将有助于发现更多潜在的治疗靶点。
3.1 DNA基因突变
目前已发现与ATC相关的突变基因,包括TP53、BRAF、RAS、ALK、PIK3CA、CTNMB1、PTEN等主要驱动基因以及几十种相关癌基因,而且这些基因在肿瘤的发展中会不断突变。在PTC中,BRAF、NRAS和HRAS基因突变的发生率分别为60.0%、8.5%和3.5%;在ATC中,BRAF、RAS和TERT启动子基因突变的发生率为47.0%、24.2%和36.4%[1,15,21]。有学者假设,ATC肿瘤的失控归因于原先存在的分化型甲状腺癌多步骤的癌基因突变[1,15]。
在ATC中最常发生的是TP53突变(57.6%),TP53突变可发生在多种不同类型的进展期癌症中,且经常发生在BRAF阳性的ATC患者中,其在ATC肿瘤发生中也有着关键的作用[15,25-26]。但是,在Landa等[15]的研究中,TP53阳性的9例ATC患者中,丝裂原激活的蛋白激酶(MAPK)途径的成分没有发生任何突变,从而支持了ATC也可以独立于已有分化型甲状腺癌(differentiated thyroid cancer, DTC)发生的假说。当肿瘤同时包含高度分化和未分化的组织学区域时,TP53突变仅限于肿瘤的未分化区域,这一观点也进一步支持了上述假说。这些分析表明,TP53失活可能直接触发分化良好的甲状腺癌细胞的肿瘤去分化或直接从正常甲状腺细胞发展为低分化型甲状腺癌或ATC。
TERT基因编码端粒酶复合物的逆转录酶成分,并在大多数癌细胞中高度表达。Sanger测序法有助于鉴定进展期甲状腺癌病例中1 295 228 C>T(C228T)和1 295 250 C>T(C250T)TERT启动子突变,据文献报道,其发生率分别为50%和33%[27]。下一代测序结果显示,TERT启动子突变的患病率实际高达73%,并且这些突变通常与其他基因改变相关。
在Landa[15]、Kunstman[27]、Jeon[28]等使用下一代测序的三项研究中,阐述了ATC的基因组和转录组情况。这些研究表明,ATC发生的独特之处在于它是由几种不同致癌性改变的累积。与以相互独立的驱动基因突变为特点的DTC相比,ATC的遗传谱通常由两个或多个突变的存在来定义。三项研究结果显示,ATC中单核苷酸变异的中位数分别为6个,4个或2个(图2)。这些研究还证实了ATC发生中新发现的驱动基因突变,其中一些突变的基因先前曾在Sanger测序研究中进行了报道,但与基于下一代测序的研究相比,它们的发生率较低。此外,由于下一代测序大大提高了检测的敏感性,鉴别出EIF1AX、MTOR、NF1、NF2、USH2A、ANKLE等几十种新发现的相关基因[1]。
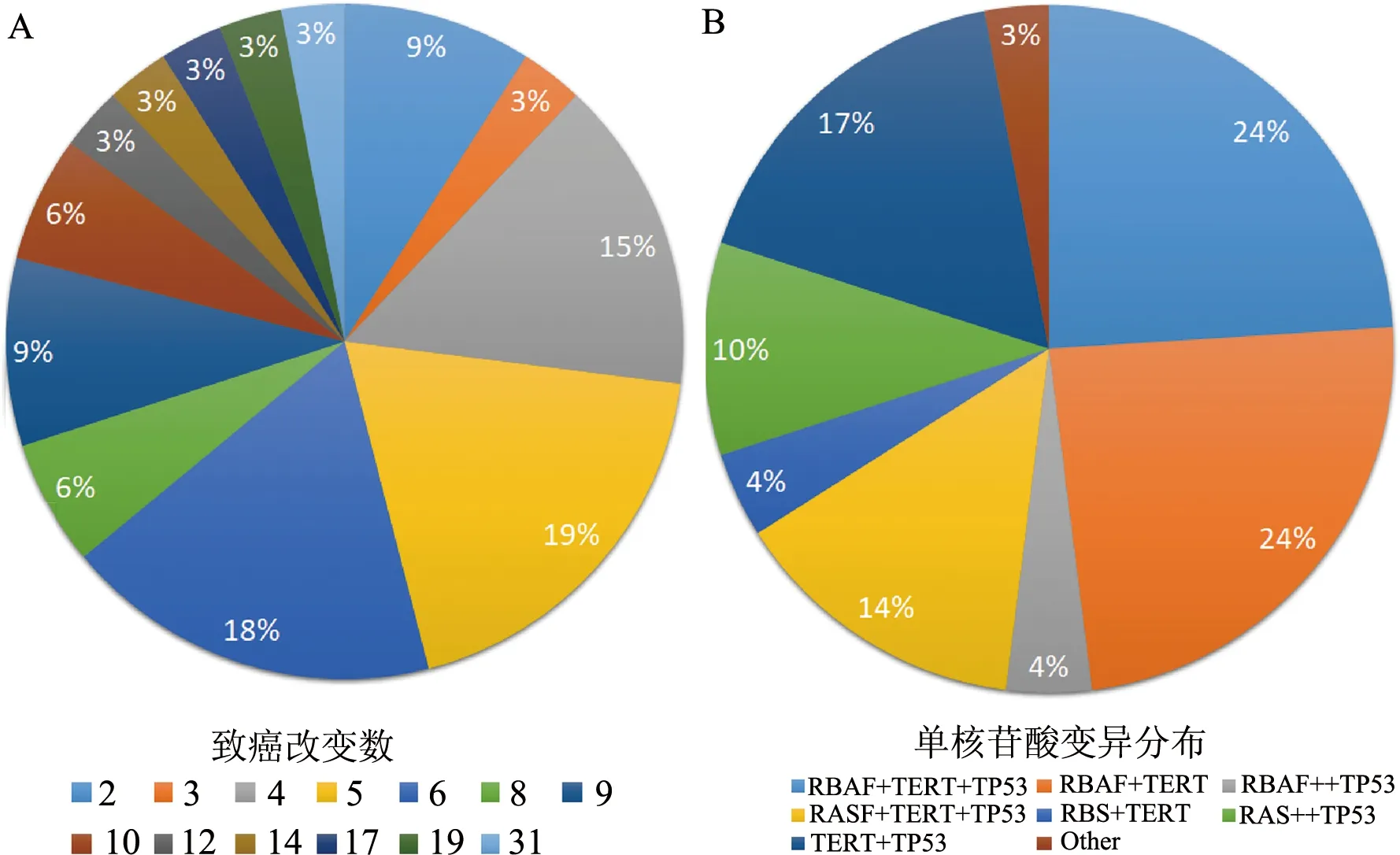
A:ATC中不同致癌性改变数量的发生率;B:ATC中多个单核苷酸变异分布的发生率,包括BRAF、TERT、RAS和其他基因
除上述基因外,真核翻译起始因子1A (EIF1AX),一种与真核翻译起始相关的基因,与DTC(尤其是滤泡状甲状腺癌)到ATC的进展有关,这可能是由于EIF1AX突变与RAS突变密切相关导致的[28]。在ATC中还发现了一些新的分子变化,例如,间变性淋巴瘤激酶(ALK)基因中的两个点突变Leu1198Phe(c.3592C>T)和Gly1201Glu(c.3602G>A)[29],这些激活的突变可以显著增加ALK的酪氨酸激酶活性,从而可以进一步上调MAPK和PI3K/AKT信号通路。此外,编码异柠檬酸脱氢酶1(IDH1)基因中的突变也与ATC的发生有关,具体机制有待进一步研究[30]。
3.2 RNA及表观遗传学改变和失调
微小RNA(miRNA)是调节转录后基因表达的非编码RNA,miRNA的调节受损在癌症发展中起着至关重要的作用。在ATC中同样也观察到miRNA表达的变化,特别是miR-146、miR-221、miR-222和miR-17-92簇等在ATC中上调,而miR-200、miR-30、let-7d和let-7g则下调。此外,miRNA表达的调控与已参与ATC发生的基因突变相关。例如,TP53突变已被证明会影响miR-200的下游激活。由于这种特定的miRNA在上皮细胞-间充质转化(epithe-lial-mesenchymal transition,EMT)的调控中起着重要作用,因此可以推测TP53突变通过控制miR-200的表达来调节EMT和肿瘤侵袭。这些分子变化代表了未来可能具有临床相关性的新疗法的潜在靶点[31]。
长链非编码RNAs(lncRNAs)是指长度大于200个核苷酸的转录本,它们没有编码蛋白质的潜力,通过异常控制基因表达的转录或转录后调控在癌症发生中发挥作用[32]。研究证实lncRNAs在甲状腺癌生物学和发病机制中发挥作用[33]。然而,有关lncRNAs在ATC发病机制中的研究较少。其中,lncRNA PTCSC3发挥肿瘤抑制因子的作用及其表达下调在ATC中得到了证实[34]。此外,lncRNAs过表达可以抑制ATC的干细胞特性和对多柔比星的耐药性。同样,lncRNA MALAT1通过调节肿瘤相关巨噬细胞分泌的成纤维细胞生长因子-2蛋白来促进甲状腺癌的血管生成[35]。
在肿瘤恶性转化的过程中,细胞会经历染色质重塑和整体表观遗传重编程,从而导致促癌基因异常激活。在这种情况下,增强的组蛋白甲基化被证实可以抑制涉及细胞周期调控、细胞黏附、凋亡和分化相关基因的功能。例如,组蛋白甲基转移酶EZH2在ATC中过表达,主要使甲状腺特异性转录因子PAX8沉默,导致侵袭性表型和较差的生存率[36]。其他受组蛋白甲基化抑制的基因还有p16、INK4A、DAPK和UCHL1[37]。除甲基化外,组蛋白去乙酰化也可以通过表观遗传,改变基因的表达。抑制整体组蛋白去乙酰化导致的分化基因的诱导,例如E-钙黏蛋白、钠碘同向转运体和甲状腺过氧化物酶等[38],有望成为一种使ATC对放射性碘治疗敏感的再分化策略。
3.3 ATC发生中的信号级联
通过将细胞周期保持在遗传毒性和非遗传毒性应激条件下,p53信号通路从根本上控制了细胞的异常生长和增殖。当发生突变或细胞质不均等分裂等任何致癌事件时,p53通路会通过诱导DNA修复、生长停滞和细胞凋亡来改变细胞命运。在ATC发病机制中,p53通路受损主要是由于TP53基因的失活突变,例如几个外显子的点突变或缺失,或者是其负调控因子如HMGA1和MDM2的过表达[37]。如前所述,BRAF突变和TP53的缺失共同促进了从PTC到ATC的进展。BRAF突变小鼠模型在敲除了p53后,均导致了ATC的进展,从而进一步证明了这一假说[39]。
MAPK通路作用在几个下游的受体酪氨酸激酶,并涉及RAS蛋白对RAF的顺序磷酸化作用,进而激活下游的MEK(MEK1、MEK2和MEK3),最终激活MAPKs,如ERK和p38。这些激活的转录因子(ERK/p38)进入细胞核并激活诸如血管内皮生长因子A、基质金属蛋白酶、抑制素、转化基因生长因子-β(TGF-β)等基因表达。这些基因通过激活细胞增殖、侵袭、迁移和血管生成来促进肿瘤发生。其中,靶基因TGF-β被证实存在于ATC的周围并促进肿瘤-基质相互作用,促进肿瘤微环境中的炎症,导致氧化应激增强,从而可以进一步激活MAPK信号传导[40]。此外,BRAFV600E介导的MAPK信号转导通过抑制经由BRAFV600E/TGF-β环的钠碘同向转运体表达来促进去分化。总之,MAPK信号参与了肿瘤-基质相互作用,并以多种方式促进了ATC的发生。
在结合生长因子后,PI3K/AKT/mTOR信号通路通过磷酸肌醇三磷酸激酶(PI3K)介导的磷酸二磷酸肌醇(PIP2)转化为磷酸三磷酸肌醇(PIP3)激活PDK1。PTEN是一种磷酸酶,可催化PIP3向PIP2的转化,从而对PI3K/PDK1/AKT信号产生负性调控。活化的PDK1依次磷酸化AKT(pAKT),后者通过磷酸化下游分子如mTOR、Bad、p21、MADD和Caspase-9发挥其生物学功能[41]。PI3K/AKT通路在滤泡状甲状腺癌中高度失调,但在滤泡状甲状腺腺瘤中却没有失调,这表明该信号通路在滤泡状甲状腺癌的发展中可能起着至关重要的作用[42]。除了持续的PI3K/AKT信号传导外,滤泡状甲状腺癌向ATC的转化还需要其他事件,例如p53失活和CTNNB1突变。
除此以外,还有一些通路在ATC中起着重要作用,并有助于肿瘤的生长和提高耐药性。例如,Wnt/β-catenin通路异常激活EMT来促进癌变;SHH通路也可以增加癌细胞的干性、运动性、迁移和EMT;同样,cMet可以激活PI3K/AKT和MAPK信号传导,从而使BRAFV600E突变的ATC对BRAF抑制剂PLX4032产生抗性[43];Janus激酶2(JAK2)是JAK家族中的一员,JAK2抑制剂来舒替尼有效减缓体外ATC细胞生长、迁移以及肿瘤的生长,表明JAK-STAT5途径在ATC发病机制中或发挥作用;AKT/GSK3β/ANG信号传导促进ATC中的血管生成;IL-11通过PI3K/AKT/GSK3β信号通路增强ATC细胞的EMT和转移[44]。
4 治疗进展
根据美国甲状腺协会指南,ATC的一线治疗包括手术切除和局部体外放射治疗。尽管采用全甲状腺切除术联合高剂量放疗可以提高生存率,但通常患者就诊时已丧失手术的机会,因此目前临床ATC的管理尚待完善,需开发新的治疗方法。
4.1 分子靶向治疗
分子靶向抑制剂作用于肿瘤特定靶分子,干扰肿瘤细胞的生长和进展,促进其凋亡的发生。这些抑制剂通常针对癌细胞信号通路中过度活跃或突变活跃的分子。近年来,靶向治疗已在众多恶性肿瘤中取得了较大的进展,例如肺癌、直肠癌、乳腺癌等[45-47]。研究表明,RAF/MAPK信号通路改变是ATC进展的重要组成部分,目前所研究的一些分子疗法包括BRAF抑制剂和MEK抑制剂均是针对这一信号级联。在单一病例的研究中,BRAF抑制剂的效果较为出色。维罗非尼(Vemurafenib)作为BRAF抑制剂中的强效型,其作用机制是通过选择性阻断BRAFV600E突变细胞的信号通路,诱导肿瘤细胞变性、凋亡。Rosove等[48]对ATC患者单独使用维罗非尼进行抑制治疗,显示单独使用维罗非尼治疗ATC疗效有限。近期进行的一项临床试验显示[49],对BRAFV600E突变的ATC患者使用达拉菲尼(150 mg/次,2次/d)和曲美替尼(2 mg/次,1次/d)联合治疗结果良好。治疗前,所有患者均接受手术或放射治疗,总反应率为69%。12个月中位反应持续时间的预测、无进展生存率和总生存率分别为90%、79%和80%。这是迄今为止第一个对ATC疾病较高临床反应的临床试验,相较于单独服用维罗非尼的疗法能够显著改善ATC患者的预后情况。
多激酶抑制剂可同时作用于两个或多个靶目标,一些潜在的多激酶抑制剂已经在临床试验中进行了测试。Baldini等[50]开展了一项关于Efatutazone联合紫杉醇应用于ATC的Ⅰ期临床研究,15例患者均接受此治疗方案,结果显示无进展生存期为3.3个月。此外,Sosa等[51]应用Fosbretabulin联合紫杉醇/卡铂治疗ATC的Ⅱ期临床试验中,纳入了8例患者,最终结果显示无进展生存期为3.3个月,总生存期为5.2个月。一项Ⅱ/Ⅲ期临床试验结果显示,联合应用紫杉醇和丙戊酸治疗ATC对总生存率和疾病进展没有任何益处[52]。
在ATC进展中,NF-κB通路发挥重要作用,卡菲佐米(Carfilzomib)作为一种有效的蛋白酶体抑制剂,可以通过上调p27和下调抗凋亡分子ATF4来靶向治疗ATC[53]。同样,硼替佐米(Bortezomib)联合MLN8054(Aurora激酶抑制剂)靶向治疗,显示ATC细胞生长减少,诱导细胞凋亡[54]。
鉴于ATC的临床特性,大多数患者会出现复发,在这些临床试验评估中,靶向治疗的疗效非常有限,仍需不断探索新的治疗方法。
4.2 免疫治疗
在ATC组织中存在肿瘤相关巨噬细胞、自然杀伤细胞和其他肿瘤浸润性淋巴细胞,突出了肿瘤-免疫细胞相互作用的相关性[55]。通过表达高水平的免疫抑制细胞因子如IL-10和TGF-β1来促进ATC的生长[56]。其他免疫抑制机制包括程序性死亡配体-1(PD-L1)与其在T细胞上表达的同源受体PD-1结合,从而下调效应T细胞功能。在ATC中,BRAFV600E突变与PD-L1的表达密切相关(P=0.015)[57]。在一项回顾性研究中,炎症细胞中PD-1的高表达(>40%染色)与较差的总生存率有关,并趋向于更差的无进展生存,而肿瘤细胞中PD-L1的高表达(>33%染色)则趋向于更差的无进展生存和总生存,表明PD-1/PD-L1通路在ATC中的关键作用[58]。在临床前的BRAFV600E/WTP53-/-动物模型研究中,BRAF抑制剂PLX4720和抗PD-L1抗体显示出显著的肿瘤消退和明显的抗肿瘤免疫反应[59]。免疫治疗的潜在疗法是用维罗非尼和纳武单抗(Nivolumab, 人IgG4 单克隆抗体)治疗含有BRAF突变和PD-L1阳性的肿瘤[60]。一些使用针对PD-1和PD-L1的抑制剂/抗体的临床试验正在ATC患者中进行。研究显示[6],另一种人源化IgG4单抗—斯巴达珠单抗(Spartalizumab)在PD-L1阳性表达和预期寿命短的患者群体中表现出良好的临床疗效和安全性,从而为PD-L1阳性进展期ATC患者(包括BRAF野生型人群)提供急需的治疗方案。与PD-1/PD-L1一样,CD70-CD27是ATC潜在的另一个关键的、与肿瘤-免疫细胞相互作用的靶点,存在于49%的ATC标本中。一项临床研究表明,CD70的表达与ATC病变中BRAFV600E突变有关,并在整个疾病进展过程中保持稳定。然而,在ATC中CD70和PD-L1之间没有观察到相关性[61]。此外,基于自然杀伤细胞的过继细胞治疗在ATC肺转移小鼠模型中显示出极佳的临床前景[62]。免疫治疗的主要障碍是肿瘤浸润性淋巴细胞数量少,因此需要探索能够改善肿瘤浸润性淋巴细胞运载的药物。
4.3 协同联合治疗
由于固有的和获得性的化学抗性,不同药物的组合经常用于临床前和临床试验以改善治疗效果。然而,在联合治疗中最重要的是建立两种药物之间的协同关系,一些研究揭示了关于药物之间组合行为的关键机制。Allegri等[63]通过证明细胞活力的丧失和EMT相关基因的下调,显示了CDK抑制剂(BP-14)和mTOR抑制剂依维莫司(Everolimus)之间的协同作用。与载体处理和多柔比星处理的小鼠相比,NF-κB抑制剂奎纳克林(Quinacrine)和索拉非尼的组合在原位小鼠模型中显示出存活率方面的改善[64]。MEK抑制剂曲美替尼联合多激酶抑制剂帕唑帕尼的研究证明,含有KRASG12R和BRAFV600E突变的异种移植肿瘤的生长显著减少[65]。BRAF抑制剂PLX4720和Src酪氨酸受体/Bcr-Abl家族抑制剂达沙替尼(Dasatinib)的组合在原位ATC小鼠模型中显示肿瘤缩小,免疫细胞浸润增加和细胞凋亡[66]。联合PPARγ配体曲格列酮和降胆固醇药物洛伐他汀,ATC细胞中表皮细胞生长因子诱导的迁移显著抑制,其本质是波形蛋白和N-钙黏蛋白的减少[67]。
相反,一些药物组合表现出非协同甚至拮抗关系。例如,将NF-κB抑制剂与紫杉烷细胞毒性药物和放射疗法组合在ATC细胞中未显示任何协同作用。类似地,Pan-MEK抑制剂U0126和BRAF抑制剂PLX4720未显示出对ATC细胞侵袭潜力的任何抑制,这表明ATC细胞中的迁移和侵袭是由其他非MEK机制介导的[68]。因此,基于肿瘤的基因组和蛋白质组学分析选择药物组合对于提高疗效至关重要。
5 结论
ATC恶性程度高、临床进展快以及预后极差,目前常规治疗的效果不佳,临床上迫切需要新的治疗手段或策略来改善ATC患者的疗效。近年来基于对ATC发生中分子机制的深入探索,ATC的治疗方案已从原来的姑息和临终关怀治疗,有望实现精准的个体化治疗(图3)。
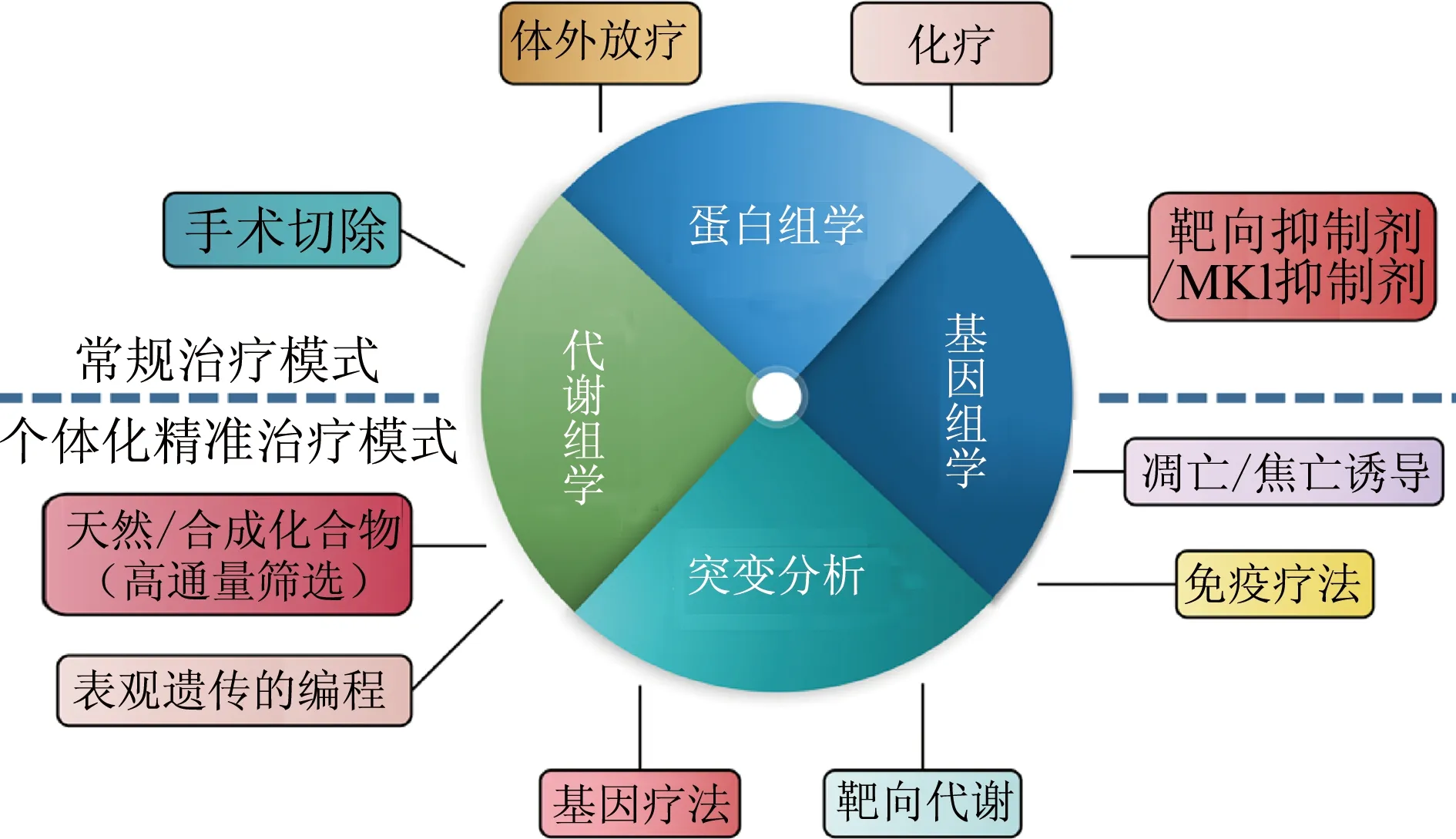
图3 ATC常规治疗模式和个体化精准治疗模式图
基于ATC致病靶点的多种精准靶向药物以及免疫治疗药物的问世,如BRAF抑制剂、多激酶抑制剂、表观遗传调节剂和凋亡诱导剂、PD-1抑制剂等给ATC患者带来了曙光,临床前试验显示出的优良疗效表明这些药物具有广阔的应用前景,多药物协同治疗有望提高ATC的治疗质量。精准医学的发展,能够从分子层次揭示ATC患者个体化的基因组学和蛋白质组学的基因突变,基于个体化致癌基因的精准分析,从而选择个体化精准治疗方案来提高患者的疗效。
