阿比特龙的研究进展
2021-03-17马宇衡
吉 庆,马宇衡
(内蒙古医科大学药学院,内蒙古 呼和浩特 010000)
前列腺癌是全球男性发病率仅次于肺癌的第二大癌症,该疾病的形成主要与患者体内雄性信号轴(ASA)有关[1-2]。患病初期由于症状不明显,因此大部分患者错过了最佳的治疗时间。目前,对于晚期前列腺癌患者最初治疗主要采用阻断体内雄激素合成疗法,但随着疾病进程的发展多数患者在接受多西他赛治疗18~24个月后,大多数前列腺癌细胞最终会发展成性激素非依赖型,患者病情会进一步演变成去势抵抗性前列腺癌(CRPC),若出现转移则会发展为转移性去势抵抗性前列腺癌(mCRPC)[3]。CRPC患者的治疗主要采用多西他赛联合泼尼松或雌二醇氮芥化疗方案[4],但由于其不良反应及耐受性,治疗效果并不理想[5]。治疗效果差主要源于上述方法并不能完全阻断雄激素的合成,有研究发现肾上腺和肿瘤本身也可以合成少量的雄激素[6],因此全面阻断体内雄激素的合成成为治疗前列腺癌的新思路。
1 阿比特龙的作用机制
研究表明,人体内孕烯醇酮及孕酮在细胞色素氧化酶P450C17(CYP17)等其他一系列酶的作用下可转化为睾酮,CYP17是该合成过程中的限速酶[7]。CYP17不仅在正常前列腺组织、睾丸及肾上腺中表达,同时前列腺癌细胞中也含有该酶,因此可将CYP17作为治疗CRPC的新靶点。阿比特龙便是基于此机制而研发的一种新型的内分泌治疗药物。
阿比特龙是由强生公司所研发,其化学名为(3β)-17-(3-吡啶基)-雄甾-5,16-二烯-3-醇,商品名为ZYTIGA。醋酸阿比特龙是阿比特龙的前体药物,经体内水解作用转化为活性代谢产物阿比特龙(图1)。阿比特龙主要是通过选择性、不可逆地抑制孕烯醇酮与孕酮转化为睾酮这一过程中的限速酶CYP17,抑制了患者体内雄激素的生成从而发挥治疗作用。相比于雄激素剥夺治疗,阿比特龙可抑制除肿瘤本身外睾丸和肾上腺中雄激素的合成,具有全面阻断雄激素合成的优点[8]。
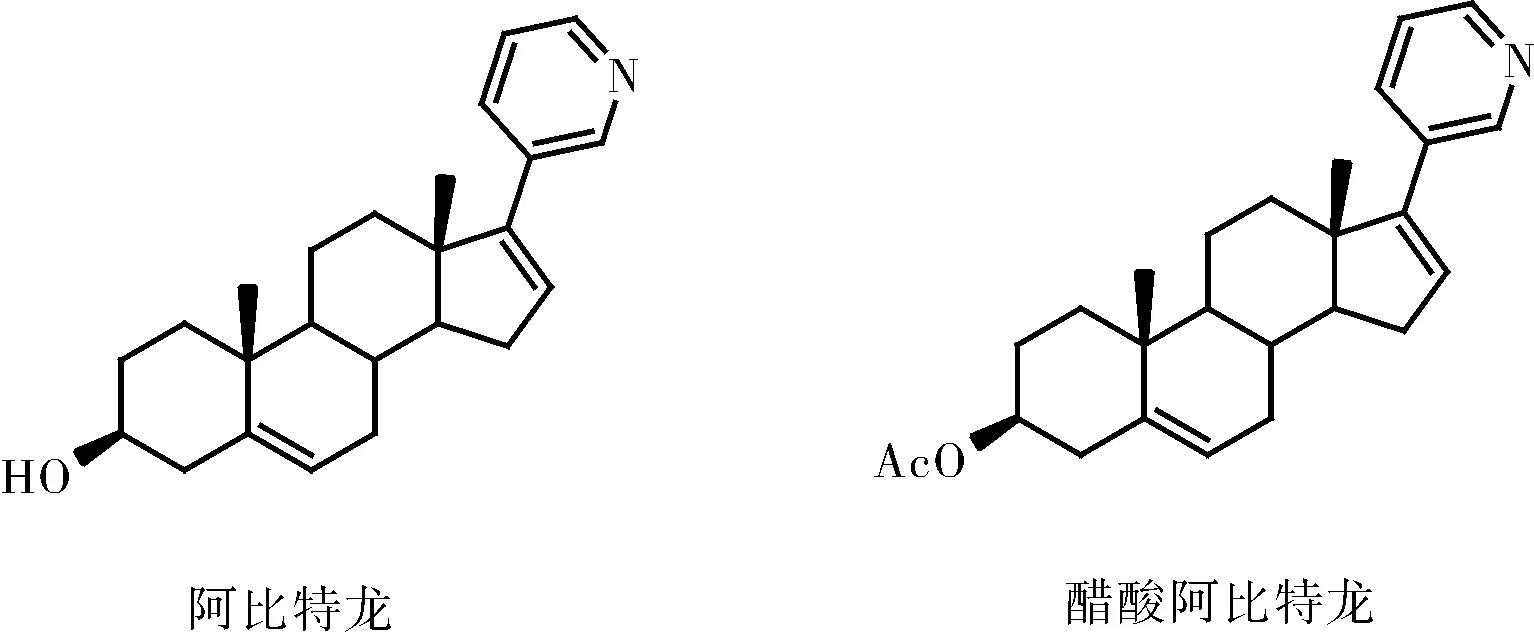
图1 阿比特龙与醋酸阿比特龙结构式
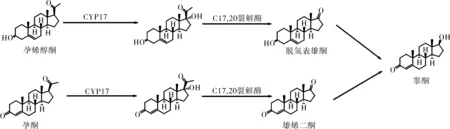
图2 阿比特龙作用机制
2 阿比特龙的临床应用与不良反应
阿比特龙于2011年4月被FDA批准上市,最初联合强的松主要用于治疗既往接受过含多西他赛化疗方案治疗后的转移性去势抵抗性前列腺癌(mCRPC)患者。III期临床试验结果表明,相比于对照组可平均延长CRPC患者生存期3.9个月[8]。之后又在2012年12月,阿比特龙被批准联合泼尼松用于未经化疗的mCRPC这一适应证[4]。由于阿比特龙抑制了CYP17从而导致会患者体内盐皮质激素积蓄以及糖皮质激素不足,服用后常会产生水肿、高血压、低血钾、水钠潴留等症状。此外,还可能会产生骨痛、尿路感染以及肝转氨酶升高等现象,所以在使用阿比特龙治疗时需与泼尼松或强的松联用,补充患者体内糖皮质激素[1]。
3 醋酸阿比特龙的合成
根据目前已有的文献报道,关于醋酸阿比特龙的制备按照起始原料的不同可分为两种方法,本文主要论述了这两种方法各自的特点,现归纳如下(图3)。
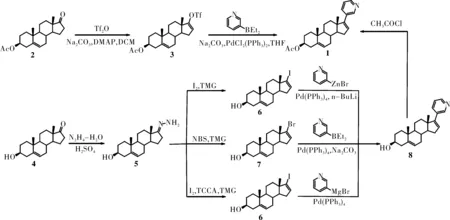
图3 醋酸阿比特龙的合成路线
3.1 以醋酸脱氢表雄酮(2)为原料
早有文献[9]报道以2为起始原料,在碱2,6-二叔丁基-4-甲基吡啶(DTBMP)作用下与三氟甲磺酸酐反应生成3,再在钯试剂PdCl2(PPh3)2催化下与二乙基(3-吡啶基)硼烷发生Suzuki偶联反应得到目标产物醋酸阿比特龙1,总产率达48.7%。但该合成过程除了要使用价格昂贵且毒性较大的三氟甲磺酸酐外,二乙基(3-吡啶基)硼烷的价格也十分昂贵,而且二乙基(3-吡啶基)硼烷的合成原料二乙基硼酸甲酯由于闪点较低,易燃易爆,运输也不方便。因此该合成途径不利于工业化生成。
3.2 以脱氢表雄酮(4)为原料
文献[10]报道以4为原料,与水合肼反应生成腙5,再在四甲基胍(TMG)存在下与碘发生碘代生成6,后与二乙基(3-吡啶基)硼烷偶联生成8,最后经乙酰化得到目标产物1,总收率为45.1%。相比于中间体3,中间体6活性较低,因此发生Suzuki偶联时80 ℃需要反应4天。反应时间的延长导致6可与4发生Heck反应生成偶联杂质,而该杂质必须借助反相色谱柱才可除去,这显然无法满足工业化生产的要求。之后王红波等[11]将二乙基(3-吡啶基)硼烷替换为3-吡啶溴化锌,将原来的反应时间缩短至室温反应5 h,总收率为56.9%。虽然该合成方法所使用的药品都比较便宜,但是3-吡啶溴化锌制备时需要在-78℃下,因此对于工业化生成还是存在一定限制。
还有文献[12]报道同样以4为原料,与水合肼生成腙5后与NBS反应生成溴代中间体7,再经Suzuki偶联及乙酰化得到目标化合物1。该合成方法虽然同样缩短了Suzuki偶联反应时间,但中间体5与NBS发生溴代时产率仅为60%,而且该合成过程同样涉及到使用价格昂贵的二乙基(3-吡啶基)硼烷,生产成本过高,不适合工业化生产。
由于3-吡啶溴化锌制备条件较为苛刻,丁亚明等[13]采用了容易制得的3-吡啶溴化镁来进行替换,之后按照文献[10]同样的方法合成了目标化合物1,总收率为51.4%。柯永新等[14]在此研究基础上,通过加入氧化剂三氯异氰脲酸(TCCA)来进行氧化碘化,总收率为54.9%。该合成方法反应条件温和,各步反应时间都较短,而且合成过程所需药品都廉价易得。同时,借助此方法可使碘单质使用量减半,减少了对环境的污染,因此此条途径比较适合工业化大规模生产。
4 结 语
综上所述,从收率、反应条件、合成路线长短、合成所需药品市场价格等方面综合考虑,文献[14]所报道的合成方法更适合企业工业化生产。
继2015年7月醋酸阿比特龙被我国批准上市后,我国于2019年7月先后批准了恒瑞与正大天晴两家药企获得醋酸阿比特龙仿制药上市资格。与此同时,美国强生公司也在积极推进其二代雄激素受体抑制剂阿帕鲁胺、阿比特龙与二代雄激素受体抑制剂的组合。因此,若想占据我国更大的前列腺癌市场,仍需研发一条更为经济实惠的合成路线。
