基于生物信息学方法构建类风湿性关节炎miRNA-mRNA调控网络*
2021-03-17曹维维袁成良
曹维维,袁成良
德阳市人民医院检验科,四川德阳 618000
类风湿性关节炎是一种累积多个系统的常见自身免疫性疾病,其主要临床表现为慢性进行性的滑膜炎症,对关节软骨和骨质造成破坏最终导致关节功能障碍。全球患病率为0.5%~1.0%[1]。类风湿性关节炎的发病机制涉及多种细胞类型,其中位于滑膜关节的成纤维样滑膜细胞在炎症、骨质破坏及血管翳的形成等病理过程中发挥重要作用[2]。研究发现,类风湿性关节炎外周血及炎症组织中的微小RNA(miRNA)存在异常表达,并与关节炎症的发生和发展、滑膜组织的增生以及滑膜细胞对凋亡作用的耐受性等病理过程密切相关[3]。本研究基于生物信息学方法构建miRNA-mRNA调控网络,为明确miRNA在类风湿性关节炎中的分子调控机制提供新的思路。
1 材料与方法
1.1材料 本研究采用美国国立生物技术信息中心(NCBI)的基因表达综合数据库(GEO数据库)获得类风湿性关节炎的基因表达谱数据。其中GSE72564包含4例类风湿性关节炎患者和4例骨关节炎滑膜样本的miRNA表达数据。GSE55235包含10例类风湿性关节炎患者和10例骨关节炎滑膜样本的mRNA表达数据。
1.2差异miRNA和差异基因的筛选 本研究将样本分为类风湿性关节炎组和骨关节炎组,应用GEO数据库中的GEO2R在线分析工具,利用t检验进行多重比较,Benjamini & Hochberg 矫正P值,设定筛选条件为|logFC|>1且P<0.05,其中FC为差异倍数(FC)。筛选得到差异表达的miRNA和差异表达基因。
1.3靶基因预测 miRNet(https://www.mirnet.ca/miRNet/upload/MirUploadView.xhtml)是一个同时收录miRTarBase v8.0,TarBase v8.0和miRecords及miRanda等多个数据库信息,具有综合分析功能的在线生物信息预测数据库。本研究采用miRNet在线数据库进行miRNA下游靶基因预测分析。
1.4功能和通路富集分析 miRNA预测的靶基因与差异表达基因取得交集,使用R语言中的clusterProfiler包对预测得到的靶基因与差异表达基因交集中的基因进行基因本体论(GO)功能富集分析及京都基因与基因组百科全书(KEGG)通路富集分析,阈值P<0.05。
1.5miRNA-mRNA调控网络构建和cytohubba分析 通过综合分析得到miRNA-mRNA关系对。利用可视化软件Cytoscape构建miRNA-mRNA调控网络图,并使用cytohubba插件进行核心基因及miRNA的筛选。
2 结 果
2.1差异表达miRNA和差异表达基因的筛选 使用GEO2R在线分析工具对miRNA和mRNA基因表达谱芯片GSE72564和GSE55235进行分析,共筛选出差异miRNA23个,其中上调miRNA6个,下调miRNA17个。共筛选到差异基因1 038个,其中643个上调基因,395个下调基因。使用ggplot包绘制火山图(图1):差异表达miRNA火山图(图1A)和差异表达基因火山图(图1B)。
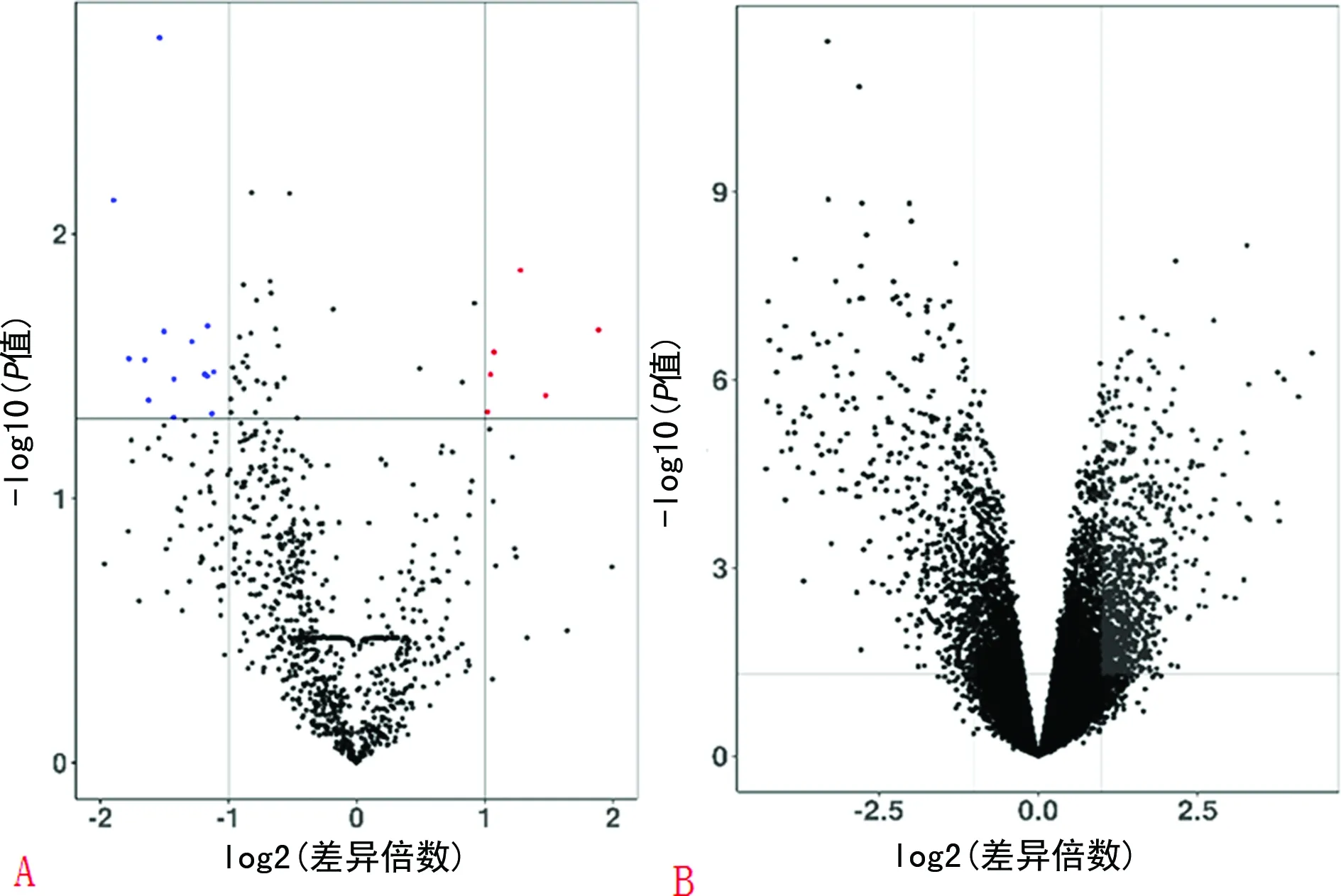
注:A为差异表达miRNA筛选;B为差异表达基因筛选。
2.2靶基因预测 分别选取下调倍数最显著的前6个miRNA:hsa-miR-30c-2-3p、hsa-miR-1305、hsa-miR-2116-5p、hsa-miR-26a-1-3p、hsa-miR-708-5p、hsa-miR-218-5p和上调的6个miRNA:hsa-miR-2276-3p、hsa-miR-1193、hsa-miR-653-5p、hsa-miR-496、hsa-miR-4263、hsa-miR-346,使用miRNet在线预测工具,对筛选出的差异miRNA进行可能的靶基因预测分析,得到2 044个预测靶基因。2 044个预测靶基因与1 038个差异基因取交集得到142个靶基因。
2.3KEGG和GO分析 GO分析:在BP(生物过程)方面,主要富集与细胞对氧含量降低的反应、缺氧反应、肌细胞分化、细胞生长调节、上皮细胞增殖、白细胞分化等生物过程。在细胞组成方面主要富集与染色外基质、染色质、转录因子复合体、细胞质膜及其蛋白复合物、受体复合物等细胞成分中。而分子功能方面主要富集与转录辅助因子活性、氨基多糖结合、染色质结合、细胞黏附分子结合、蛋白异源二聚化活性等分子功能。KEGG分析:主要富集与磷脂酰肌醇3-激酶/蛋白激酶B(PI3K/AKT)信号通路、人类T细胞白血病病毒Ⅰ型感染、癌症中的转录失调、EB病毒感染、乙型肝炎、库欣综合征、Th17细胞分化、Th1/Th2细胞分化等信号通路。
2.4miRNA-mRNA调控网络的构建和cytohubba分析 使用cytoscape(v3.8.0)可视化软件绘制miRNA-mRNA的调控网络图,见图2。三角形节点表示miRNA,矩形节点代表mRNA。图2中可见一个靶基因可受多个miRNA调控,而1个miRNA则可调控多个靶基因。其中hsa-miR-218-5p调控的靶基因最多且以上调为主,其次为hsa-miR-30c-2-3p,同样以上调为主。使用cytohubba插件,通过最大团中心性MCC算法筛选出前十位的hub基因:PPP2R1B、MGAT4C、TUBB2A、ZNF124、CAPZB、HMGA1、HNRNPA1、HSPG2、KLHL21、TOP2A、WARS、CRISPLD2。
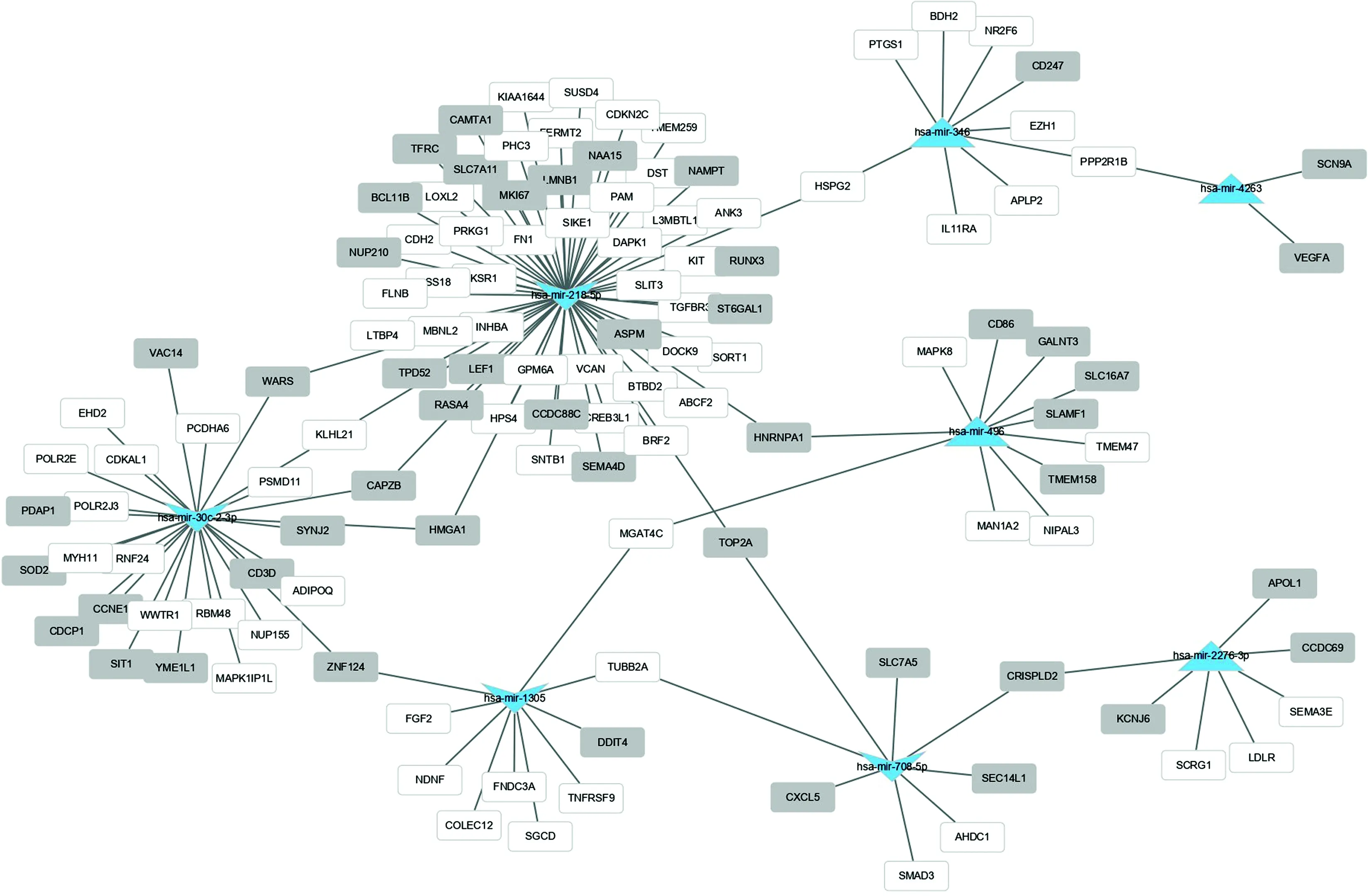
注:■为下调的基因;□为上调的基因;▲为上调的miRNA;▼为下调的miRNA。
3 讨 论
类风湿性关节炎作为一种累积多个系统的常见自身免疫性疾病。研究发现遗传易感性、病毒感染、环境及生活方式等是类风湿性关节炎目前已知的危险致病因素[4-5]。miRNA在类风湿性关节炎患者的血液、关节、滑膜和不同细胞类型中均存在差异表达,而这些差异表达的miRNA可通过增强或抑制不同细胞的增殖分化、迁移和侵袭及血管新生等病理过程参与介导疾病的发生和发展。本研究以miRNA的作用机制为基础,通过生物信息分析方法成功构建miRNA-mRNA调控网络,进一步探讨miRNA及相关信号通路在类风湿性关节炎发生和发展中的分子调控机制,为类风湿性关节炎的靶向诊断和治疗提供可靠的理论依据。
利用GEO数据库中的GSE72564和GSE55235,筛选与类风湿性关节炎相关的差异表达的miRNA和mRNA,共筛选出差异miRNA23个(上调miRNA6个和下调miRNA17个)及差异表达基因1 038个(643个上调基因和395个下调基因)。通过对预测靶基因与差异表达基因进行整合,筛选出142个候选基因。进行KEGG通路富集和GO功能富集分析,发现其主要富集于缺氧反应及细胞黏附分子结合等生物过程和分子功能,参与调控PI3K/AKT信号通路、Th17细胞分化、Th1/Th2细胞分化等信号通路。
类风湿性关节炎的滑膜组织炎症及成纤维样滑膜细胞(FLS)的大量增殖和分化使耗氧量增加,导致局部缺氧。缺氧诱导因子1α(HIF-1α)作为缺氧诱导因子HIF家族成员之一,对氧水平敏感并在缺氧条件下显著增加。低氧环境可以使HIF-1α的积累,驱动血管内皮生长因子(VEGF)及多种促血管生成介质如CXCL8、CCR20的表达[6-7]。还可以增加促炎因子如白细胞介素(IL)-6、金属基质蛋白酶的分泌及增加滑膜细胞的侵袭性[8]。同时研究证实,多种细胞黏附分子与类风湿性关节炎的发生和发展密切相关[9-10]。MELINTE等[11]发现,类风湿性关节炎患者的滑膜软骨组织中含有大量的血小板内皮细胞黏附分子1(PECAM-1/CD31)。CD31作为免疫球蛋白超家族的成员,位于内皮细胞的细胞间边界上,在炎症、细胞凋亡及白细胞的跨内皮迁移中起到关键作用[12]。此外,研究者通过类风湿性关节炎患者的FLS的共培养实验结果显示,钙黏蛋白11在FLS细胞间的接触点上表达,与α-连环蛋白和β-连环蛋白共定位形成黏附连接[13],进而证明了钙黏蛋白11在FLS细胞间的黏附作用。
PI3K/AKT信号通路作为一种重要的细胞内信号转导通路,已被证实与类风湿性关节炎的发生发展有关[14]。有研究表明,类风湿性关节炎患者FLS中的PI3K/AKT信号通路可以通过刺激细胞因子如IL-17、IL-22等引起FLS细胞的异常增殖并加重滑膜炎性反应[15]。此外,PI3K/AKT信号通路可通过刺激Th17细胞分泌IL-17使FLS表达过量的核因子κB受体活化因子配体(RANKL),促进破骨细胞的分化和形成从而导致骨质破坏。值得一提的是,Th17细胞及幼稚CD4+T细胞的分化一定程度上也与PI3K/AKT信号通路的调节有关[16]。
Th17作为CD4+T细胞的亚群,可分泌IL-17A、IL-17F和IL-21等多种细胞因子,在类风湿性关节炎患者外周血、关节滑液和滑膜组织中广泛表达并参与促进炎性反应和软骨破坏等多种病理过程。研究表明类风湿性关节炎患者存在Th1/Th2失衡,Th1亚群占优势,Th1通过分泌IL-2和γ-干扰素等细胞因子参与细胞免疫,Th2通过分泌IL-4/IL-10等细胞因子参与体液免疫[17]。Th1的异常增加导致促炎细胞因子的产生,加重炎症和组织损伤的发生[18-20]。
本研究利用人类基因表达芯片,采用差异基因分析和靶基因预测等生物信息分析方法,构建了miRNA-mRNA网络。hsa-miR-218-5p、has-miR-30c-3p、hsa-miR-1305、hsa-miR-708-5p表达下调,而hsa-miR-496、hsa-miR-346、hsa-miR-2276-3p、hsa-miR-4263表达为上调。研究发现,hsa-miR-218-5p是成骨分化的重要诱导因子,其可通过靶向结合分泌型糖蛋白Slit及其跨膜受体Robo蛋白(Slit-Robo)途径的跨膜受体蛋白1(Robo1)并抑制Dickkopf-1(DKK-1)的分泌从而促进类风湿性关节炎的FLS成骨分化[21]。长链非编码RNA DANCR在滑膜间充质干细胞软骨形成过程中起关键作用,hsa-miR-1305作为的DANCR的下游靶点,其过度表达可以通过降低转化生长因子-β通路成员Smad4的表达进而抑制hsa-miR-1305的表达而诱导软骨细胞的增殖和分化作用[22]。hsa-miR-708-5p可通过抑制Wnt3a蛋白/细胞内β-连环蛋白(Wnt3a/β-catenin)通路在蛋白和转录水平的活性促进细胞凋亡并抑制细胞增殖从而参与类风湿性关节炎的疾病调节机制[23]。SEMAAN等[24]发现,hsa-miR-346可利用RNA结合蛋白稳定作用控制类风湿性关节炎中肿瘤坏死因子-α蛋白的释放及其mRNA的稳定性。
在Hub基因中,KLHL21编码一组高度保守的蛋白,涉及多种细胞和分子并广泛参与到炎性反应、氧化应激反应等病理过程。研究证实,类风湿性关节炎患者滑膜巨噬细胞中的KLHL21表达上调[25]。MEI等[26]发现,KLHL21可靶向作用于核转录因子-κB(NF-κB)抑制蛋白激酶β(IKKβ),对肿瘤坏死因子-α(TNF-α)激活的NF-κB信号通路产生负性调节作用,其过表达可以诱导细胞因子IL-8、IL-1β等多种促炎因子的释放致使炎症加重。硫酸肝素蛋白多糖2可以编码一种大的多结构域蛋白Perlecan,作为血管外基质的重要组成部分,它有助于维持内皮屏障功能,也是一种有效的平滑肌细胞增殖抑制剂,有助于维持血管稳态。Perlecan可以和多种细胞表面受体,如整合素、细胞外基质分子和生长因子如成纤维细胞生长因子、内皮生长因子、血小板衍生生长因子等相互作用,从而在介导细胞的迁移、增殖分化等过程中发挥重要作用[27-28]。hsa-miR-218-5p作为高频下调表达的miRNA,可同时靶向作用于KLHL21和 HSPG2的表达并下调二者水平。因此该miRNA-mRNA调控网络与类风湿性关节炎的发病机制间重要关系可作为新的研究方向进行深入探究。
综上,本研究通过差异基因筛选及靶基因预测等生物信息学分析方法,成功构建了miRNA-mRNA调控网络并进行信号通路及功能富集分析和揭示了与类风湿性关节炎相关的关键基因。这些关键基因在类风湿性关节炎的发生发展过程中起到重要作用,为后续深入类风湿性关节炎的诊断和治疗提供了新的诊断靶点和治疗思路。
