Development of a Microfluidics-Based Quantitative Real-Time PCR to Rapidly Identify Photobacterium damselae subsp. damselae with Different Pathogenicity by Detecting the Presence of mcp or dly Gene
2021-03-06ZHANGZhengYUYongxiangCHENJingWANGYingengJIANGYongLIAOMeijieRONGXiaojunandZHANGHao
ZHANG ZhengYU YongxiangCHEN JingWANG Yingeng*,JIANG YongLIAO MeijieRONG Xiaojunand ZHANG Hao
Development of a Microfluidics-Based Quantitative Real-Time PCR to Rapidly Identifysubsp.with Different Pathogenicity by Detecting the Presence oforGene
ZHANG Zheng1),2), YU Yongxiang1), CHEN Jing1), WANG Yingeng1),2), *,JIANG Yong3), LIAO Meijie1),2), RONG Xiaojun1),2), and ZHANG Hao1)
1),,,,266071,2),,266237,3),266071,
As a marine bacterial pathogen,subsp.(PDD) is distributed in seawater worldwide. It can infect different animals as well as humans, even cause deaths. The highly conserved regions of PDDgene on chromosome andgene on plasmid were selected as the target fragments to design the specific primers. Recombinant plasmid standard was prepared based on the primers. With GENECHECKER UF-150 qRT-PCR instrument as the platform, a fluorescence-based quantitative real-time PCR (qRT-PCR) method was established for the detection of PDD. This method can specifically detect PDD and distinguish the highly virulent strains. Additionally, the test results can be qualitatively judged by visualization, while the quantitative detection can be achieved through the standard curve calculation. The minimum limit of detection was 1.0×101copiesμL−1, and the detection time was less than 20min. In summary, this new method has outstanding advantages, such as strong specificity, high sensitivity, and low site requirements. It is a rapid on-site detection technology for highly virulent PDD strains.
mariculture;; microfluidics; pathogenicity; rapid detection
1 Introduction
subsp.(PDD) is a subspecies ofbelonging tofa- mily. As an important pathogen of marine animals, it is widely distributed in the seawater worldwide. The bacterium is firstly reported in 1981, which was isolated from skin ulcers of damselfish (), and ori- ginally was named as(Love., 1981). Subsequently, there are successive reports on PDD infection in different animals around the world, including fish (Ketterer., 1992; Labella., 2006), crustaceans (Song., 1993; Vaseeharan., 2007), mollusks (Han-lon., 1984; Lozano-León., 2003), sea turtles (Oben-dorf., 1987), mammals (Fujioka., 1988; Lee., 2018).In 1982, the first case of PDD infection in human was reported (Morris., 1982). Symptoms of human PDD infection usually appear as severe fasciitis (Yuen., 1993) and septicemia (Perez-Tirse., 1993). In 2004, Japanese scientists reported two cases of human PDD in- fection, causing severe fasciitis to acute death (Yamane., 2004). In 2015, a child was infected with PDD and died in Saudi Arabia (Alhemairi., 2015). These cases have confirmed that the bacteria are highly pathogenic to humans.
Usually PDD is studied with heterogeneously isolated strains according to their phenotypic characteristics and virulence (Pedersen., 1997; Terceti., 2016; Ter- ceti., 2018). Some of them show strong hemolytic activity and high pathogenicity, while others exhibit weak hemolytic activity and lower pathogenicity, or even no pa- thogenicity. Early studies have confirmed that the pathogenic strains of PDD can produce a large amount of cytolytic toxin, which is named as damselysin (Dly) (Ko- thary., 1985; Kreger., 1987; Cutter., 1990). Subsequently, more researches revealed that a pore-form- ing toxin with hemolytic activity, named as phobalysin P (PhlyP), is another important virulence factor of the strain(Rivas., 2011, 2013a, 2015; Vences., 2017). These two toxins are encoded bygene andplgene, respectively, which are located on the virulence plasmid named as pPHDD1 (Rivas., 2011). In 2013, a study reported the third virulence gene on chromosome encoding another hemolysin (HlyA) (Rivas., 2013a). To date, adequate studies have supported that the conjunction of Dly, PhlyP and HlyA cytotoxins constitutes the virulence system of highly virulent lineages of PDD (Rivas., 2015; Rivas., 2013b). In fact, different strains of the same bacteria with significant differences in pathogeni- city are common in nature (Saroj., 2008; Cheng., 2019).
Until now, infections of mariculture animals by PDD have been rarely reported, and PDD is considered as a newly discovered pathogen in China. Nevertheless, according to available studies, it can infect different marine fishes living in different environments, such as(Zhang., 2009),(Yan., 2018),(Zhang., 2019b) and so on. There seems to be no obvious regional specificity of this bacterium though the seawater temperatures in different environments are quite different. In 2016, we isolated a PDD strain from black rockfishwith the symptom of skin ulceration, which were cultured in net-cage of Changdao County, Shandong Province. The experiment has confirmed its pathogenicity to black rockfish (Zhang., 2019b). Its whole-genome sequencing has also been completed (Yu., 2019). Together with other 24 strains isolated previously, there are totally 25 PDD strains in our laboratory. These strains have significantly different physiological and biochemical characteristics as well as hemolytic activity and drug resistance (Shi., 2019). When 108cfumL−1live bacterial suspension was injected into the fish, it can cause the death of experimental fish in 2 days, while no death was observed in the fish injected with non-pathogenic strain.In addition, our previous research has confirmed that PDD is widely distributed along China offshore seawater, pos- ing a potential public health risk to fishermen, offshore workers and tourists. Therefore, in order to control PDD infection in animals and humans, it is necessary to estab- lish a rapid detection method to distinguish these strains with different levels of virulence.
2 Material and Methods
2.1 Experimental Strains
A total of 20 strains belonging to different bacterial spe- cies and 25 different strains of PDD were selected in this study. The 20 different bacterial species included three, three, two, two, two, two, one, one, one, onesubsp.(PDP) and two. Among them, seven strains of,,,,,andwere purchased from Chi- na General Microbiological Culture Collection Center (CG- MCC) as standard strains. The remained 13 strains were respectively isolated from different diseased animals in the laboratory of Yellow Sea Fishery Research Institute (YSFRI). More detailed information of these 20 strains was listed in Table 1. The genomic sequences of two strains were detected and submitted to NCBI (Yu., 2019; Zhang., 2019a).

Table 1 The 23 bacteria strains for primers specific detection of PDD mcp gene fragment
Notes: CGMCC, China General Microbiological Culture Collection Center. YSFRI, Yellow Sea Fishery Research Institute.
All the 25 PDD strains (Fig.4) were isolated from different diseased marine animals along China coastline. They were all stored in YSFRI. These strains showed significant differences in terms of hemolytic phenotype and pa- thogenicity to black rockfish (Shi., 2019). Two of these strains, Pdd1601 (α-hemolysis) and Pdd1605 (β-he- molysis), had completed whole-genome sequencing data. One of them has been submitted to NCBI (Yu., 2019).
2.2 Specific Primer Design
Based on the whole-genome sequencing data of two strains, Pdd1601 and Pdd1605, a highly conserved sequence ingene with a length of 348bp and a highly conserved sequence ingene with a length of 695bp were respectively selected as the targets to design specific primers. Thegene was used to distinguish PDD from other different bacteria, and thegene was used to distinguish PDD strains with different pathogenicity. Specific primers were designed through Primer Premier 5.0 software and synthesized in Sangon Biotech (Shanghai) Co., Ltd. (Shanghai, China). Thegene primer sequences were as follows: (5’–3’) TGAAATTGCCCAACTGTCCC (forward) and TCACTTACTTGGGCCACATC (reverse). Thegene primer sequences were as follow: (5’–3’) TTTGGACGAGCGGTCCATTT (forward) and GGAG CCCAATCTTGACCAGG (reverse).
2.3 Preparation of Standards
The 348-bp target fragment ingene was amplified using the designed primers, and the PCR product was li- gated to PMD18-T vector, and transformed into DH-5α competent cells. The positive clones were enriched to ex- tract plasmids, which were then digested and identified through sequencing. The correctly recombined plasmid was named Pl-Pdd and used as the positive standard in the present study. The copy number of recombinant plasmids was calculated using a previously established method (Ray- mond., 2004). Briefly, the OD260nmwas firstly deter- mined to calculate the plasmid concentration. Then the co- py number was calculated according to the formula as follows:

2.4 Establishment of qRT-PCR System and Standard Curve
The genomic DNA of bacterial strains was extracted using TIANamp Bacterial DNA kit (TIANGEN Biotech Co., Ltd., Beijing, China). The qRT-PCR was conducted using Realplex model quantitative real-time PCR instrument (Eppendorf Co., Ltd., Hamburg, Germany) in a 20- μL reaction system consisting of 10μL of 2×SYBR Green Pro Taq HS PREMIX (Takara Biomedical Technology Co., Ltd., Beijing, China), 0.4μL of 10μmolL−1gene for- ward primer, 0.4μL of 10μmolL−1gene reverse pri- mer, 0.2μL of 20μmolL−1ROX Reference Dye, 2μL of DNA template and 7μL of RNase-free water. Briefly, after an initial denaturation step at 95℃ for 30s, the amplifications were carried out with 40 cycles at a melting temperature of 95℃ for 10s, an annealing temperature 60℃ for 20s and an extension temperature of 72℃ for 20s, followed by a melting curve analysis (95℃ for 15s, 60℃ for 15s, 60℃ to 95℃ for 20min, 95℃ for 15s).
The prepared standard plasmid Pl-Pdd was diluted into different gradient concentrations using 10-fold serial dilution, and a qRT-PCR was performed according to the optimized reaction conditions to draw a standard curve.
2.5 The Primer Specificity and Universality
The specificity of designed primers ofgene fragment was verified by qRT-PCR under the optimized conditions using the genomic DNA of 23 bacterial strains (Ta- ble 1) as the template, and RNase-free water was used as a negative control.
The universality ofgene primers was verified using the genomic DNA of 25 PPD strains as the template, and RNase-free water was used as a negative control.
The specificity of designed primers ofgene fragment was verified by qRT-PCR using the genomic DNA of 25 PPD strains as template, and RNase-free water as a negative control.
2.6 The Sensitivity of mcp Gene Primers
The standard plasmid Pl-Pdd was serially diluted intoa concentration gradient consisting of 1.0×101–1.0×107copiesμL−1using 10-fold serial dilution, which was used as templates. The primer sensitivity was verified by qRT- PCR under the reaction conditions in Section 2.4. Meanwhile, ordinary PCR was carried out to compare the sensitivity for designed primers ofgene.
2.7 Establishment of a Microfluidics-Based Real- Time PCR Method for PDD Detection
GENECHECKERUF-150 Ultra-Fast qRT-PCR instru- ment (CHK Biotech Co., Ltd., Shanghai, China) was used to establish a microfluidics-based qRT-PCR method for PDD detection. The PCR was conducted in a 10-μL reac- tion system consisting of 5μL of 2×ChamQ Universal SYBR qPCR Master Mix (Vazyme Biotech Co., Ltd., Nan- jing, China), 1μL of 10μmolL−1forward primer, 1μL of 10μmolL−1reverse primer, 2μL of bacterial DNA template and 1μL of RNase-free water. Briefly, after an initial de- naturation step at 95℃ for 30s, the amplifications were carried out for 30 cycles with a melting temperature of 95℃ for 5s, an annealing temperature of 60℃ for 20s, and an extension temperature of 72℃ for 5s, followed by melt- ing curve analysis (95℃ for 5s, 60℃ for 40s, and 95℃ for 5s). Fluorescence detection was performed after the qRT-PCR was finished. The amplified products using spe- cific primers ofandgenes in different channels of the chip were required by synchronous detection.
3 Results
3.1 The Recombinant Plasmid Plmcp-Pdd
In the present study, we successfully amplified the target sequence with 348bp in length using the Pdd1601 strain DNA as template and-F/R primer pairs. The PCR product was recovered and ligated, and the construct- ed recombinant standard plasmid Pl-Pdd (Fig.1)was tran- sformed into DH-5α competent cells. The recombinant plas- mid was used as the positive standard, and its concentra- tion was detected as 67.70ngμL−1, which was converted into 2.03×1010copiesμL−1.
3.2 Amplification Curve and Standard Curve of the Plasmid Plmcp-Pdd
The standard plasmid Pl-Pdd was diluted to 2.03× 102–2.03×108copiesμL−1. Using it as a template, the amplification was performed under the optimized qRT-PCR conditions mentioned in section 2.4. The amplification cur- ve (Fig.2I) and standard curve (Fig.2II) were obtained based on the results, indicating that there was a good linear rela- tionship between the amount of Pl-Pdd plasmid copies and Ct value. The formula was=−3.012+36.70, and the correlation coefficient2was 0.998.
Fig.1 Verification of ligation and transformation of PDD mcp gene target fragment with electrophoresis. M, DNA molecular quality standards (DL2000); 1, 2, Enzyme-di- gested products.

Fig.2 Amplification curve and standard curve of standard plasmid Plmcp-Pdd. I, Amplification curve of different concentrations of plasmid Plmcp-Pdd. A, 2.03×108copiesμL−1; B, 2.03×107copiesμL−1; C, 2.03×106copiesμL−1; D, 2.03×105copiesμL−1; E, 2.03×104copiesμL−1; F, 2.03×103copiesμL−1; G, 2.03×102copiesμL−1. II, Standard curve of plasmid Plmcp- Pdd.
3.3 Specificity and Universality of the Designed Primers
The designed primers forgene were used to amplify the DNA fragments of 23 strains listed in Table 1. The results showed that only three PDD strains were successfully amplified. The target sequence was about 348bp in length (Fig.3), confirming the specificity of the designed primers to PDD strains.
Moreover, qRT-PCR was performed to amplify the plas- mid Pl-Pdd and the other 10 strains using the designed primers forgene. These 10 strains were chosen from Table 1, including(VR-01),(VH-01),(VS-01),(VP-01),(VAl-01),(AF- 01),(VA0531),(VC0406), PDP (Pdp1810) and(EC-01). The amplification curve was only observed from the plasmid Pl-Pdd, while negative results were found in other 10 strains. The results reconfirmed the specificity of the designed primers to PDDgene.

Fig.3 Specific detection of the designed primers for PDD mcp gene fragment. M, DNA molecular quality standards (DL2000); 1, V. rotiferianus (VR1601); 2, V. rotiferianus (VR1602); 3, V. rotiferianus (VRST-01); 4, PDD (Pdd0905); 5, PDD (Pdd1601); 6, PDD (Pdd1605); 7, V. harveyi (VHST-01); 8, V. harveyi (VH0207); 9, V. harveyi (VH1809); 10, V. anguil- larum (VA1012); 11, V. anguillarum (VA0531); 12, V. splendidus (VSST-01); 13, V. splendidus (VS1805); 14, V. para- haemolyticus (VPST-01); 15, V. parahaemolyticus (VP0531); 16, V. alginolyticus (VAlST-01); 17, V. alginolyticus (VAl 1811); 18, A. fischeri (AFST-01); 19, V. scophthalmi (VSc0531); 20, V. cyclitrophicus (VC0406); 21, PDP(Pdp1810); 22, E. coli (EC0701); 23, E. coli (EC ST-01); 24, RNase-free H2O.
Amplification results of the 25 PDD strains showed that all these strains harbored the target fragment (Fig.4), de- monstrating the good intraspecific universality of the de- signedgene primers in PDD strains.
The 25 PDD strains were amplified with the designedgene primers. The results showed that the target fragment of 695bp in length was only detected in two strains with high hemolytic activity (Fig.5), confirming the good specificity of the designed primers to PDDgene.

Fig.4 Intra-specific universality detection of the designed primers for PDD mcp gene fragment. M, DNA molecular quali- ty standards (DL2000); 1, Pdd0210; 2, Pdd0905; 3, Pdd0906; 4, Pdd0907; 5, Pdd0908; 6, Pdd0909; 7, Pdd1208; 8,Pdd1308; 9, Pdd1605; 10, Pdd1606; 11, Pdd1607; 12, Pdd1608; 13, Pdd1609; 14, Pdd1611; 15, Pdd1612; 16, Pdd1613; 17, Pdd1614; 18, Pdd1615; 19, Pdd1616; 20, Pdd1617; 21, Pdd1701; 22, Pdd1706; 23, Pdd1807; 24, Pdd1809; 25, Pdd1808; 26, RNase-free H2O.
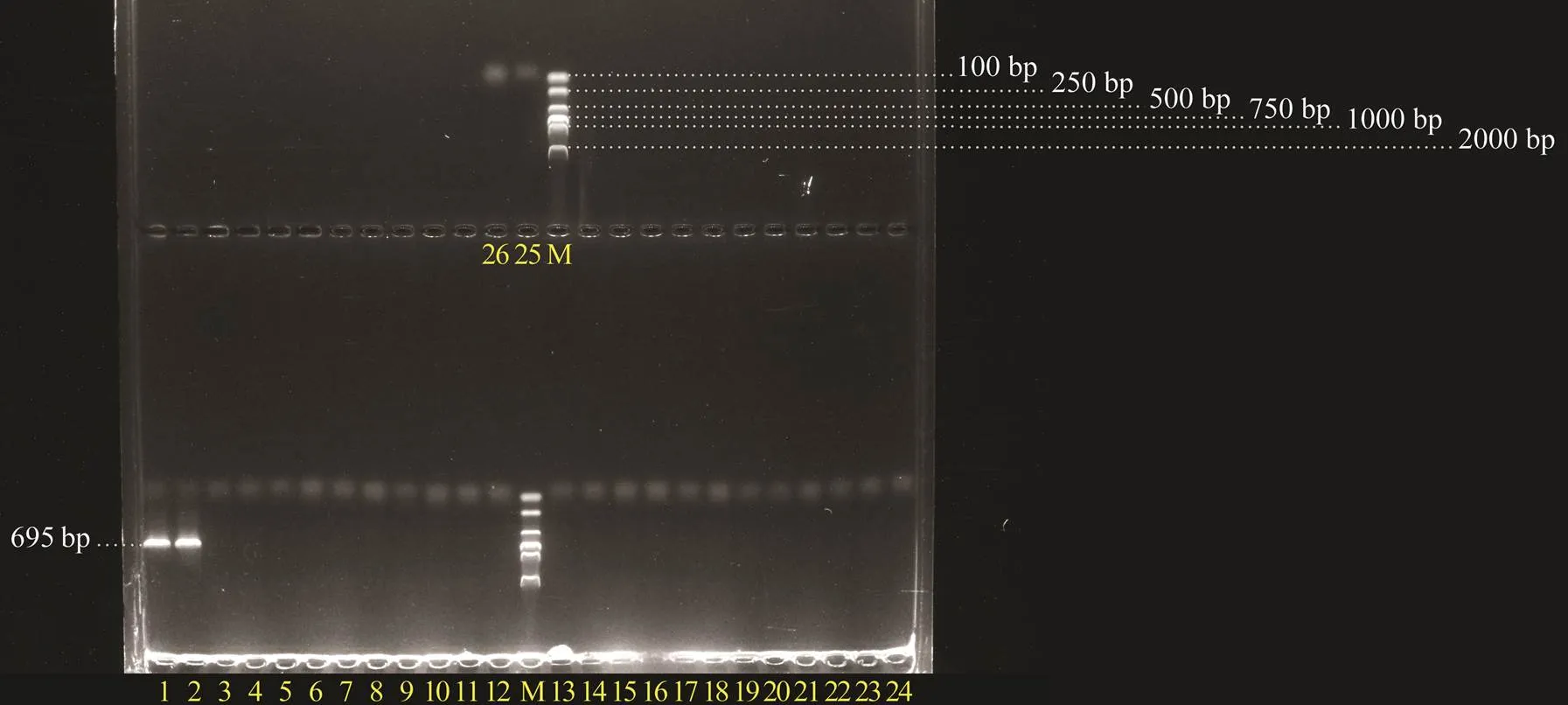
Fig.5 Specific detection of the designed primers for PDD dly gene fragment. M, DNA molecular quality standards (DL- 2000); 1, Pdd1605; 2, Pdd1608; 3, Pdd0210; 4, Pdd- 0905; 5, Pdd0906; 6, Pdd0907; 7, Pdd0908; 8, Pdd0909; 9, Pdd1208; 10, Pdd1308; 11, Pdd1606; 12, Pdd1607; 13, Pdd1609; 14, Pdd1611; 15, Pdd1612; 16, Pdd1613; 17, Pdd1614; 18, Pdd1615; 19, Pdd1616; 20, Pdd1617; 21, Pdd1701; 22, Pdd1706; 23, Pdd1807; 24, Pdd1809; 25, Pdd1808; 26, RNase-free H2O.
3.4 The Sensitivity of mcp Gene Primers
Comparing the results of qRT-PCR and ordinary PCR amplifications using the designedgene primers show- ed that the detection limit of qRT-PCR was as low as 1.0× 101copiesμL−1, while it was 1.0×104copiesμL−1for ordinary PCR (Fig.6). The sensitivity of qRT-PCR was 1000 times higher compared with the ordinary PCR.

Fig.6 Detection sensitivity comparison of qRT-PCR and ordinary PCR using the primersof mcp gene. I, Amplification curve of qRT-PCR; II, Gel detection of ordinary PCR; A, 1.0×107copiesμL−1; B, 1.0×106copiesμL−1; C, 1.0×105copiesμL−1; E, 1.0×104copiesμL−1; E, 1.0×103copiesμL−1; F, 1.0×102copiesμL−1; G, 1.0×101copiesμL−1; M, DNA molecular quality standards (DL2000); 1, 1.0×105copiesμL−1; 2, 1.0×105copiesμL−1; 3, 1.0×103copiesμL−1; 4, 1.0×102copiesμL−1; 5, 1.0×101copiesμL−1; 6, RNase-free H2O.
3.5 The Microfluidics-Based qRT-PCR Detection
The microfluidics-based ultra-fast PCR method for PDD detection was established using the specific primers ofgene andgene under the optimized reaction condi- tions, and GENECHECKER UF-150 was used as the ins- trument platform. Into two channels in the chip,gene primers andgene primers added respectively. After the PCR amplification, detection results were visually judged through fluorescence signal. If fluorescence signal simul- taneously appeared in both two channels, the test object could be confirmed as the highly pathogenic PDD strain. If fluorescence signal appeared in the channel withgene primers and no fluorescence signal was detected in the channel withgene primers, the test object could be confirmed as the low or non-pathogenic PDD strain. If no fluorescence signal was detected in both two channels, or if fluorescence signal was only detected in the channel withgene primers, the test object was not a PDD strain (Fig.7). The reaction time of qRT-PCR was reduced to about 17min. To obtain the Ct value in the amplification curve based on the standard curve, the quantitative detec- tion of PDD strains was performed. The lowest limit detection was 1.0×101copiesμL−1, with extremely high sensitivity. Due to the outstanding portability of the instrument, this method could be used for field testing.
4 Discussion
As a pathogenic bacterial strain, which is widely distributed in the marine environment worldwide, PDD is also an important zoonotic pathogen. PDD has no obvious host specificity, and it can infect not only poikilotherm, but also homotherm (Rivas., 2013), or even cause acute death of humans (Yamane., 2004). Sufficient attention has to be paid to this pathogen. From the existing studies, there are changeable phenotypes among different PDD strains. They show diversities in pathogeni- city, hemolysis, antibiotic resistance, physiological and bio-chemical characteristics (Takahashi., 2008). Environ-mental stress factors, such as temperature and salinity, canalso affect the virulence of PDD strains (Vences., 2017;Matanza., 2018). Our previous research has also con- firmed that the strains isolated from the same mariculture environment have significantly different pathogenicities to black rockfish,. Therefore, it is important to quickly detect the PDD strains with different pathogeni- cities to prevent and control the infections of such bacteria.
The classical method of bacterial detection usually uses 16Sgene (Cole., 2009) andgene (Sun., 2019). Many bacterial PCR-based classification and identification methods have been successfully established according to these genes. However, it is difficult to accu- rately distinguish PDD species due to the extremely high homology of16S. For example, the homology betweenandis 99.6%, while it is 99.8% betweenand(Wen., 2009). Thegene also needs to cooperate with other genes to achieve higher inter-spe- cies discrimination (Teh., 2010). The highly virulent plasmid pPHDD1 encoding damselysin (Dly) and phoba- lysin P (PhlyP), two key virulence factors of highly viru- lent PDD strains, has been reported in 2011 (Rivas., 2011). Moreover, the third hemolysin encoding gene ofchlocated on PDD chromosome has been found in 2013 (Rivas., 2013). From the existing researches,andplon the plasmid pPHDD1 andchon the chromosome constitute the virulence system of PDD together. Their synergy and participation of some functional genes create the high pathogenicity and strong hemolytic activity. The absence or silence of key virulence factorscan cause a significant decrease of pathogens’ virulence (Rivas., 2013; Rivas., 2015; Luo., 2019). It offers a new possible method for distinguishing PDD strains with different virulence through detecting the existence of virulence factors or functional genes.

Fig.7 Detection results of different bacterial strains using GENECHECKER UF-150 qRT-PCR instrument. The chip has 10 reaction channels, of which the 1st, 3rd, 5th and 7th channels are added with mcp gene primers, the 2nd, 4th, 6th and 8th channels are added with dly gene primers, and the 9th and 10th channels are added with RNase-free H2O. In chip I, the 1st, 2nd, 3rd, 5th and 7th channels appear fluorescence, 1st and 2nd channels are highly pathogenic PDD strain (Pdd1605), 3rd and 4th channels are lowly pathogenic PDD strain (Pdd0906), 5th and 6th channels are non-pathogenic PDD strain (Pdd0909), 7th and 8th channels are lowly pathogenic PDD strain (Pdd0210), 9th and 10th channels are RNase-free H2O. In chip II, the 1st, 2nd, 3rd, 5th and 7th channel appear fluorescence, 1st and 2nd channels are highly pathogenic PDD strain (Pdd1608), 3th and 4th channels are lowly pathogenic PDD strain (Pdd1208), 5th and 6th channels are lowly pathogenic PDD strain (Pdd1611), 7th and 8th channels are lowly pathogenic PDD strain (Pdd1616), 9th and 10th channels are RNase-free H2O. In chip III, the 1st, 2nd and 3rd channel appear fluorescence, 1st and 2nd channels are highly pathogenic PDD strain (Pdd1605), 3rd and 4th channels are lowly pathogenic PDD strain (Pdd1809), 5th and 6th channels are PDP (Pdp1810), 7th and 8th channels are V. parahaemolyticus (VPST-01), and 9th and 10th channels are RNase-free H2O.
In the present study, we collected 25 different PDD strains along the coast of China. These strains exhibited significant differences in pathogenicity to black rockfish. We com- pleted the whole-genome sequencing of two strains, which displayed notable difference in hemolytic phenotype and pathogenicity. From the results, bothgene on plasmid andgene on chromosome were selected together as target sequences for PDD detection. Thegene was employed to distinguish PDD from other different bacte- ria, while thegene was used to identify PDD strains with high pathogenicity. It has been confirmed that Dly is one of the most important virulence factors encoded by plasmid pPHDD1, which can vanish with the loss of the plasmid. Methyl-accepting chemotaxis proteins (Mcps) are the most common receptors widely found in bacteria and archaea. They not only participate in various physiologi- cal activities of cells, but also play an important role in pathogenicity of many bacteria (Ud-Din., 2017). Geneis usually considered to be a housekeeping gene,while its sequence greatly varies in different species. Through genome-wide analysis, we screened out thegene of PDD and found that this gene was ubiquitous in PDDstrains with excellent specificity. It can be employed to distinguish PDD from other strains, including subsp., and can be used as a target gene for PDD detection. Moreover, such gene might also be a potential indicator to identify the two subspecies of.
Notably, the two strains that were detected to harborgene showed strong hemolytic activity on sheep blood plate and high pathogenicity to black rockfish, whereas the other stains withoutgene exhibited weak hemolytic ability and low or non-pathogenicity. From these results, we speculated thatgene is directly associated with the strong hemolytic activity and high pathogenicity of PDD. The sequence ofgene in this study was derived from a megaplasmid (374kb) of a highly virulent PDD strain, which was much larger than the 153-kb virulent plasmid pPHDD1. This finding confirmed the existence of other highly virulent plasmids in addition to pPHDD1 in PDD.
Another aim of this study was to establish a microfluidics-based qRT-PCR technique for the detection of PDD strains. As an emerging technology of molecular biology, microfluidics-based PCR, with the advantages of short- time consumption and low cost, has been widely used in medical application and research areas (De Paz., 2014;Park., 2011). We successfully established a microfluidics-based qRT-PCR method for PDD detection through specific primers. The test results can also be qualitatively judged by visualization. Quantitative detection would be achieved through the standard curve calculation. The mi- nimum detection limit was as low as 1.0×101copiesμL−1, and the detection time was shortened to less than 20min. This method has the feasibility of on-site detection, which is suitable to develop fast and accurate PDD detection for field research.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2019YFD0900 104), and the Key Projects of Science and Technology In- novation of Shandong Province (No. 2018YFJH0703).
Alhemairi, M., Alghanmi, F., and Alshamrani, A. S., 2015. Child death due to infection withsubs., a new case., 23 (3): 176-178.
Cheng, G., Hussain, T., Sabir, N., Ni, J., Li, M., Zhao, D., and Zhou, X., 2019. Comparative study of the molecular basis of pathogenicity ofstrains in a mouse model., 20 (1): 5.
Cole, J. R., Wang, Q., Cardenas, E., Fish, J., Chai, B., Farris, R. J., Kulam-Syed-Mohideen, A. S., McGarrell, D. M., Marsh, T., Garrity, G. M., and Tiedje, J. M., 2009. The ribosomal da- tabase project: Improved alignments and new tools for rRNA analysis., 37 (Sup l): D141-D145.
Cutter, D. L., and Kreger, A. S., 1990. Cloning and expression of the damselysin gene from., 58 (1): 266-268.
De Paz, H. D., Brotons, P., and Muñoz-Almagro, C., 2014. Mole- cular isothermal techniques for combating infectious diseases: Towards low-cost point-of-care diagnostics., 14 (7): 827-843.
Fujioka, R. S., Greco, S. B., Cates, M. B., and Schroeder, J. P., 1988.from wounds in bottlenose dolphins., 4 (1): 1-8.
Hanlon, R. T., Forsythe, J. W., Cooper, K. M., Dinuzzo, A. R., Folse, D. S., and Kelly, M. T., 1984. Fatal penetrating skin ul- cers in laboratory-reared octopuses., 44 (1): 67-83.
Ketterer, P. J., and Eaves, L. E., 1992. Deaths in captive eels () due to()., 69 (8): 203-204.
Kothary, M. H., and Kreger, A. S., 1985. Purification and char- acterization of an extracellular cytolysin produced by., 49 (1): 25-31.
Kreger, A. S., Bernheimer, A. W., Etkin, L. A., and Daniel, L. W., 1987. Phospholipase D activity ofcytolysin and its interaction with sheep erythrocytes., 55 (12): 3209-3212.
Labella, A., Vida, M., Alonso, M. C., Infante, C., Cardenas, S., Lopez-Romalde, S., Manchado, M., and Borrego, J. J., 2006. First isolation ofssp.from cultured redbanded seabream,Valenciennes, in Spain., 29 (3): 175-179.
Lee, K., Kim, H. K., Sohn, H., Cho, Y., Choi, Y. M., Jeong, D. G., and Kim, J. H., 2018. Genomic insights intosubsp.strain KC-Na-1, isolated from the finless porpoise ()., 37: 26-30.
Love, M., Teebken-Fisher, D., Hose, J. E., Farmer, J. J., Hickman, F. W., and Fanning, G. R., 1981., a marine bacterium, causes skin ulcers on the damselfish., 214 (4525): 1139-1140.
Lozano-León, A., Osorio, C. R., Nuñez, S., Martínez-Urtaza, J., and Magariños, B., 2003. Occurrence ofsubsp.in bivavlve molluscs from Northwest Spain., 23 (1): 40-44.
Luo, G., Xu, X., Zhao, L., Qin, Y., Huang, L., Su, Y., and Yan, Q., 2019.is a key virulence gene duringinfection., 42 (7): 991-1000.
Matanza, X. M., and Osorio, C. R., 2018. Transcriptome changes in response to temperature in the fish pathogensubsp.: Clues to understand the emer- gence of disease outbreaks at increased seawater temperatures., 13 (12): e0210118.
Morris, J. G., Wilson, R., Hollis, D. G., Weaver, R. E., Miller, H. G., Tacket, C. O., Hickman, F. W., and Blake, P. A., 1982. Illness caused byand., 319 (8284): 1294-1297.
Obendorf, D. L., Carson, J., and McManus, T. J., 1987.infection in a stranded leatherback turtle ()., 23 (4): 666-668.
Park, S., Zhang, Y., Lin, S., Wang, T. H., and Yang, S., 2011. Ad- vances in microfluidic PCR for point-of-care infectious di- sease diagnostics., 29 (6): 830-839.
Pedersen, K., Dalsgaard, I., and Larsen, J. L., 1997.associated with diseased fish in Denmark., 63 (9): 3711-3715.
Perez-Tirse, J., Levine, J. F., and Mecca, M., 1993.: A cause of fulminant septicemia., 153 (15): 1838-1840.
Raymond, C. R., and Wilkie, B. N., 2004. Th-1/Th-2 type cytokine profiles of pig T-cells cultured with antigen-treated mo- nocyte-derived dendritic cells., 22 (8): 1016-1023.
Rivas, A. J., Balado, M., Lemos, M. L., and Osorio, C. R., 2011. Thesubsp.hemolysins damselysin and HlyA are encoded within a new virulence plasmid., 79 (11): 4617-4627.
Rivas, A. J., Balado, M., Lemos, M. L., and Osorio, C. R., 2013a. Synergistic and additive effects of chromosomal and plasmid- encoded hemolysins contribute to hemolysis and virulence insubsp.., 81 (9): 3287-3299.
Rivas, A. J., Lemos, M. L., and Osorio, C. R., 2013b.subsp., a bacterium pathogenic for marine animals and humans., 4:Article 283.
Rivas, A. J., Vences, A., Husmann, M., Lemos, M. L., and Osorio, C. R., 2015.subsp.major virulence factors Dly, plasmid-encoded HlyA, and chro- mosome-encoded HlyA are secreted via the Type II scretion system.,83 (4): 1246-1256.
Rivas, A. J., Von Hoven, G., Neukirch, C., Meyenburg, M., Qin, Q., Füser, S., Boller, K., Lemos, M. L., Osorio, C. R., and Hus- mann, M., 2015. Phobalysin, a small β-pore-forming toxin ofsubsp.., 83 (11): 4335-4348.
Saroj, S. D., Shashidhar, R., Karani, M., and Bandekar, J. R., 2008. Distribution of Salmonella pathogenicity island (SPI)-8 and SPI-10 among different serotypes of Salmonella., 57 (4): 424-427.
Shi, L. N., Yu, Y. X., Jiang, Y., Zhang, Z., Wang, Y. G., Liao, M. J., and Rong, X. J., 2019. Studies on the phenotypic differences of differentsubsp.strains., 43 (6): 15-24 (in Chinese with English abstract).
Song, Y. L., Cheng, W., and Wang, C. H., 1993. Isolation and characterization of, infectious for cultured shrimp in Taiwan., 61 (1): 24-31.
Sun, Y., Zhuang, Z., Wang, X., Huang, H., Fu, Q., and Yan, Q., 2019. Dual RNA-seq reveals the effect ofgene ofon immune response of., 87: 515-523.
Takahashi, H., Miya, S., Kimura, B., Yamane, K., Arakawa, Y., and Fujii, T., 2008. Difference of genotypic and phenotypic characteristics and pathogenicity potential ofsubsp.between clinical and environmen- tal isolates from Japan., 45 (2): 50- 158.
Terceti, M. S., Ogut, H., and Osorio, C. R., 2016.subsp., an emerging fish pathogen in the Black Sea: Evidence of a multiclonal origin., 82 (13): 3736-3745.
Terceti, M. S., Vences, A., Matanza, X. M., Dalsgaard, I., Pe- dersen, K., and Osorio, C. R., 2018. Molecular epidemiology ofsubsp.outbreaks in ma-rine rainbow trout farms reveals extensive horizontal gene trans- fer and high genetic diversity., 9: Ar- ticle 2155.
The, C. S. J., Chua, K. H., and Thong, K. L., 2010. Simultaneous differential detection of human pathogenic and nonpathogenicspecies using a multiplex PCR based onB andA genes., 108 (6): 1940-1945.
Ud-Din, A. I. M. S., and Roujeinikova, A., 2017. Methyl-accept- ing chemotaxis proteins: A core sensing element in prokaryotes and archaea., 74 (18): 3293-3303.
Vaseeharan, B., Sundararaj, S., Murugan, T., and Chen, J. C., 2007.ssp.associated with dis- eased black tiger shrimpFabricius in India., 45 (1): 82-86.
Vences, A., Rivas, A. J., Lemos, M. L., Husmann, M., and Osorioa, C. R., 2017. Chromosome-encoded hemolysin, phospho- lipase, and collagenase in plasmidless isolates ofsubsp.contribute to virulence for fish., 83 (11): e00401- 17.
Wen, W. Y., Xie, Z. Y., Xu, X. D., Zhang, X. Z., Zhang, S. X., and Zhou, Y. C., 2009. Establishment of a rapid PCR detection method forbased ongene., 28 (10): 575-578 (in Chinese with English abstract).
Yamane, K., Asato, J., Kawade, N., Takahashi, H., Kimura, B., and Arakawa, Y., 2004. Two cases of fatal necrotizing fasciitis caused byin Japan., 42 (3): 1370-1372.
Yan, N., Zhang, Z. Q., Wu, T. L., Zhu, J. X., Fu, Y. F., Han, H. S., Wang, H. B., Shi, Q. M., and Gao, G. S., 2018. Isolation and identification ofsubsp.(PDD) from Tongue Sole., 10 (2): 99-114.
Yu, Y. X., Zhang, Z., Wang, Y. G., Liao, M. J., Rong, X. J., Li, B., Wang, K., Chen, J., and Zhang H., 2019. Complete ge- nome sequence ofsubsp.strain SSPD1601 isolated from deep-sea cage-culturedwith septic skin ulcer., 2019: Article 4242653.
Yuen, K. Y., Ma, L., Wong, S. S. Y., and Ng, W. F., 1993. Fatal necrotizing fasciitis due to., 25 (5): 659-661.
Zhang, X. J., Qin, G. M., Chen, C. Z., Fang, G., and Yan, B. L., 2009. Biological characterization and phylogenetic analysis ofsubsp.from diseasedL., 30 (3): 38-43 (in Chinese with English abstract).
Zhang, Z., Yu, Y. X., Jiang, Y., Wang, Y. G., Liao, M. J., Rong, X. J., Wang, K., Zhang, H., and Chen, J., 2019a. First report of isolation and complete genome ofstrain SSVR1601 from cage-cultured black rockfish () associated with skin ulcer., 42 (5): 623-630.
Zhang, Z., Yu, Y. X., Wang, K., Wang, Y. G., Jiang, Y., Liao, M. J., and Rong, X. J., 2019b. First report of skin ulceration caused bysubsp.in net-cage cul- tured black rockfish ()., 503: 1-7.
March 27, 2020;
May 8, 2020;
August 25, 2020
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2021
. E-mail: wangyg@ysfri.ac.cn
(Edited by Qiu Yantao)
杂志排行

Journal of Ocean University of China的其它文章
- Corrosion Mechanism of 5083 Aluminum Alloy in Seawater Containing Phosphate
- In vitro Antioxidant Effects of Porphyra haitanensis Peptides on H2O2-Induced Damage in HepG2 Cells
- Molecular Characterization and Expression Analysis of SKIV Infection of Interferon-Induced Protein with Tetratricopeptide Repeats 1 (IFIT1) in Epinephelus lanceolatus
- Characteristics and Influencing Factors of the Microbial Concentration and Activity in Atmospheric Aerosols over the South China Sea
- Characteristics of Atmospheric Rivers over the East Asia in Middle Summers from 2001 to 2016
- Otolith Shape Analysis as a Tool to Identify Two Pacific Saury (Cololabis saira) Groups from a Mixed Stock in the High-Seas Fishing Ground