氯化锂对Aβ1-42引起的星形胶质细胞谷氨酸释放的抑制作用及其对海马神经元损伤的保护作用
2021-01-31张显晨李香雨林钰淼杨颜齐闫恩志
刘 琪, 张显晨, 李香雨, 林钰淼, 杨颜齐, 闫恩志
(锦州医科大学基础医学院药理学教研室,辽宁 锦州121001)
阿尔茨海默病(Alzheimer disease,AD)越近晚期疗效越差,研究[1-3]表明:抑制谷氨酸的兴奋性损伤,在AD 早期的防治中具有重要意义,但目前尚缺乏有效的治疗药物及作用靶点。在脑组织中,星形胶质细胞(astrocytes,AS)是谷氨酸代谢的关键场所,不仅参与谷氨酸的代谢循环,还对细胞外高浓度谷氨酸具有缓冲调节作用[4]。近年来 的 研 究[5]证 实: β 淀 粉 样 蛋 白(β-amyloid protein,Aβ)可激活AS 释放谷氨酸,引起神经元损伤以及突触结构和功能的改变。长期以来锂盐是治疗双相障碍(躁狂抑郁症)的有效药物。临床研究[6-7]显示:锂可降低双相障碍患者晚期并发AD的概率;临床和动物实验[8-10]证明:长期使用小剂量锂制剂,可延缓AD 患者认知功能障碍的进展;最近的研究[11-12]显示:锂盐可对抗急性刺激所致AS 的老化损伤,而AS 的老化可促进谷氨酸对神经元的兴奋性损伤,表明锂可能在AD 早期具有预防和治疗作用,但其机制目前还不清楚。本研究前期实验[13-14]证实:氯化锂可抑制细胞膜去极化所致AS 内质网上三磷酸肌醇(inositol triphosphate,IP3)和雷尼丁(Ryanodine)敏感性内Ca2+储池及谷氨酸的释放,但该作用是否对Aβ 引起的AS 谷氨酸的释放具有抑制作用尚未见研究报道。本研究通过离体培养AS 和海马神经元的方法,探讨氯化锂的对β 淀粉样蛋白1-42(β-amyloid protein 1-42,Aβ1-42)所致神经元损伤的保护作用,并阐明其机制,为AD 的治疗及新药的开发提供依据和治疗靶点。
1 材料与方法
1.1 药品和试剂氯化锂、谷氨酸、L-多聚赖氨酸、二甲基亚砜(DMSO)、二丁酰环腺苷酸(dBcAMP)、硝苯地平(Nifedipine)、毒胡萝卜素(Thapsigargin) 和Aβ1-42购 于 美 国Sigma 公 司,Ryanodine和钠-钙交换蛋白1(Na+-Ca2+exchanger 1,NCX1) 抑 制 剂KB-R7943 购 于 美 国Calbiochem 公司,Fura-2 AM 和DMEM 培养基购于美国Invitrogen 公司,马血清购于广州蕊特生物科技有限公司,瞬时受体电位通道蛋白1 (transient receptor potential channel 1,TRPC1)、NCX1、电压门控钙离子通道蛋白(CACNA1C,Cav1.2)一抗和山羊抗兔二抗均购自美国Santa Cruz 公司,β-肌动蛋白(β-actin)购于上海碧云天试剂公司,一氧化氮(nitric oxide,NO)检测试剂盒购于北京邦定生物工程公司,SuperSignal West Pico 化学发光底物购于美国Thermo scientific 公司。
1.2 AS 培养AS 培养参见本课题组前期采用的方法[13-14],取出生1 d 的CD-1 小鼠大脑皮层。剥离脑膜后,将脑组织切碎,震荡1 min,用100 μm 和70 μm 滤膜过滤,将细胞悬液接种于培养皿中用于谷氨酸释放实验检测,或将细胞悬液接种于经过多聚赖氨酸处理的盖玻片上用于钙离子水平检测。培养基为含10 %马血清的DMEM。培养3 周后用于实验,加入0.25 mmol·L-1dBcAMP 促进AS 分化成熟。
1.3 海马神经元培养及分组取出生24 h 内的小鼠乳鼠,75%酒精浸泡乳鼠消毒1 min,断头,置于无菌DMEM 培养基内,无菌条件,剥离大脑,取出海马组织,置于DMEM/F12 培养基中,剪成1 mm×1 mm×1 mm 的小块。加入0.125%胰蛋白酶37 ℃消化10 min。细胞分散后,加入少量10%胎牛血清终止消化。200 目筛网过滤,1 000 g 离心10 min,细胞悬液由含有10%胎牛血清和10%马血清的DMEM/F12 培养基制成,调整细胞密度为1×105L-1~1×107L-1,接种在铺有L-多聚赖氨酸培养皿或6 孔细胞培养板中,置于37 ℃、5%CO2培养箱中培养。培养48 h 后加入含阿糖胞苷(终浓度为5 mg·L-1) 的培养基,抑制非神经元增殖。以后每3 d 半量换液1 次,7 d 后用于实验。海马神经元成熟化后,分为3 组:①对照组,不进行处理;②Aβ1-42组,AS 培养体系内加入250 nmol·L-1Aβ1-42作用48 h,将Aβ1-42处理后的AS 条件培养基(ACM)加入海马神经元中作用24 h;③Aβ1-42+不同剂量氯化锂组,向预先加入0.25、0.50 和1.00 mmol·L-1氯 化 锂 处 理 后 的ACM 中 加 入250 nmol·L-1Aβ1-42作用48 h,将处理后的ACM加入培养的海马神经元中作用24 h。各组以Aβ1-42处理48 h 的不含AS 的条件培养基为对照。
1.4 荧光探针技术检测AS中Ca2+水平盖玻片上进行原代AS 培养,AS 分为对照组、 Aβ1-42+不同剂量(0.25、0.50 和1.00 mmol·L-1) 氯化锂组和 阻 断 剂 组 (Nifedipine 组、KB-R7943 组 和Ryanodine 组),Aβ1-42+不同剂量氯化锂组加入不同剂量氯化锂作用2 周。AS 在37oC 培养箱中,加入含Fura-2 AM(5 μmol·L-1)的等渗液进行荧光负载30 min,反复冲洗细胞2 次,去除没有进入细胞荧光染 料。等 渗 液(mmol·L-1):137 NaCl,5 KCl,0.44 KH2PO4,4 NaHCO3,1.3 CaCl2,0.8 MgSO4,0.5 MgCl2·6H2O,10 Glucose。将细胞置于荧光显微镜下,调整参数为20 s 测定一个间隔循环。每孔加入等渗液180 μL,待曲线平稳后,加入终浓度为250 nmol·L-1Aβ1-42刺激细胞,各阻断剂组在加入Aβ1-42前,分别向细胞中加入100 nmol·L-1Nifedipine、10 μmol·L-1KB-R7943 和1 μmol·L-1Ryanodine 各20 μL 作用10 min。在激发波长分别为340 和380 nm、发射波长为510 nm 条件下,进行双波长细胞中Ca2+水平测定。实验测定60 个循环。 Ca2+再充功能实验中,AS 分为对照组和Aβ1-42+0.50 mmol·L-1氯化锂组,按上述方法荧光负载后,每孔加入180 μL 无Ca2+等渗液,置于荧光显微镜下,待曲线平稳后,向细胞中加入20 μL终浓度为1 μmol·L-1的Thapsigargin 作用10 min 后,再加入终浓度为2 mmol·L-1的CaCl2,测定40 个循环。荧光检测结果以波长340 和380 nm 荧光的比值(F340/F380)表示。
1.5 谷氨酸释放实验AS 分为对照组、Aβ1-42组和Aβ1-42+不同剂量(0.25、0.50和1.00 mmol·L-1)氯化锂组,氯化锂组加入不同剂量氯化锂作用2 周后,去除培养皿中的培养液,加入缓冲液清洗30 min,去除缓冲液,置于台面上倾斜15°,用蠕动 泵 以0.5 mL·min-1流 速 加 入250 nmol·L-1的Aβ1-42刺激细胞,收集细胞上清液,取400 μL 上清液加入200 μL 邻苯二甲醛(ortho-phthalaldehyde,OPA) 溶 液 衍 生3 min,进 行 高 效 液 相 色 谱(HPLC)程序,计算谷氨酸释放量,用1 mol·L-1的氢氧化钠收集细胞,进行蛋白测定,将测定值代入标准曲线公式,计算出每分钟、每毫克细胞释放的谷氨酸量。
1.6Western blotting 法检测AS 中钙离子通道蛋白表达水平AS 分为对照组和Aβ1-42+不同剂量(0.25、0.50 和1.00 mmol·L-1) 氯化锂组,处理方法见“1.5”,将培养皿中AS 置于冰盒上,加入裂解液后收集细胞,进行蛋白定量,蛋白浓度调成一致后进行SDS-聚丙烯酰胺凝胶电泳,分离的蛋白经过转膜后封闭,加入一抗后4℃过夜,二抗孵育2 h(1∶1 000 稀释),进行ECL 化学发光,图像采集,实验重复3 次后采用Visionworks 6.3.3 分析软件对蛋白条带进行分析,以TRPC1、NCX1 和Cav1.2 蛋白条带灰度值与β-actin 条带灰度值之比表示目的蛋白表达水平。
1.7 海马神经元树突形态学观察体外培养海马神经元分组及处理见“1.3”。采用配备CCD 和Tsview 软件的Olympus CKX41SF 型倒置相差显微镜,通过10 倍或20 倍物镜观察,确保让一个神经元拍摄在一个视野中,通过40 倍物镜观察树突分支,以保证能够清楚展现全部树突分支。分析时采用Ohara 和Havton 命名法:从胞体直接伸出的树突分支为一级分支,从一级分支伸出的为二级分支,依次类推。用SPOT 软件测量随机选取的海马神经元,测量树突分支总长度(total dendrite branch length,TDBL)、 神 经 元 一 级 树 突 分 支 数(primary-order dendrite number,PDN) 和神经元树突总分支数(maximum branch order,MAO)。计数样本从3 个不同批次培养的神经元获取,每批次随机选择10~15 个细胞,采用双盲方法。
1.8 Griess 法检测海马神经元培养液中NO 释放量海马神经元分组及处理见“1.3”。用1 mol·L-1亚硝酸钠溶液配制标准品(0、1、2、5、10、20、40、60 和100 μmol·L-1)。按50 μL/孔在96 孔细胞培养板中加入标准品和样品。按50 μL/孔,在每个孔中分别加入室温Griess Reagent Ⅰ和Griess Reagent Ⅱ。在波长540 nm处检测吸光度(A)值。将亚硝酸钠浓度(μ mol·L-1) 作为横坐标,测得A 值作为纵坐标,绘制标准曲线,计算回归方程及相关系数,吸取各组培养海马神经元的培养上清液,测定NO 释放量。
1.9 统计学分析采用SPSS 17.0 统计软件进行统计学分析。所有实验结果来自于单独培养的3 个批次的细胞,各组细胞中Ca2+水平,谷氨酸释放量,细胞中TRPC1、NCX1 和Cav1.2 蛋白表达水平,海马神经元TDBL、PDN 和MBO,海马神经元NO 释放量,均符合正态分布,以表示,多组间样本均数比较采用单因素方差分析,组间均数两两比较采用LSD 法。以α=0.05 为检验水准。
2 结 果
2.1 加入不同阻滞剂后各组AS中Ca2+水平培养于盖玻片上成熟的AS 用Fura-2 AM 处理后进行细胞内Ca2+测定,Fura-2 AM 可穿透细胞膜进入细胞内与Ca2+结合,结合Ca2+后在340 nm 激发光下可以产生较强的荧光(绿色),而在380 nm 激发光下则会导致荧光减弱,以F340/F380 表示细胞中Ca2+水平(图1)。采用250 nmol·L-1Aβ1-42刺激,能持久引起AS 中Ca2+水平升高,分别应用L-channel 钙通道抑制剂Nifedipine、Na+-Ca2+抑制剂KB-R7943 和Ryanodine 受体抑制剂Ryanodine后,细胞中Ca2+水平升高均被抑制,而Na+-Ca2+抑制剂KB-R7943 并不能阻断细胞中Ca2+水平升高(图2)。

图1 荧光显微镜下观察各组AS 中Ca2+水平(×200)Fig.1 Levels of Ca2+in AS in various groups observed under fluorescence microscope(×200)
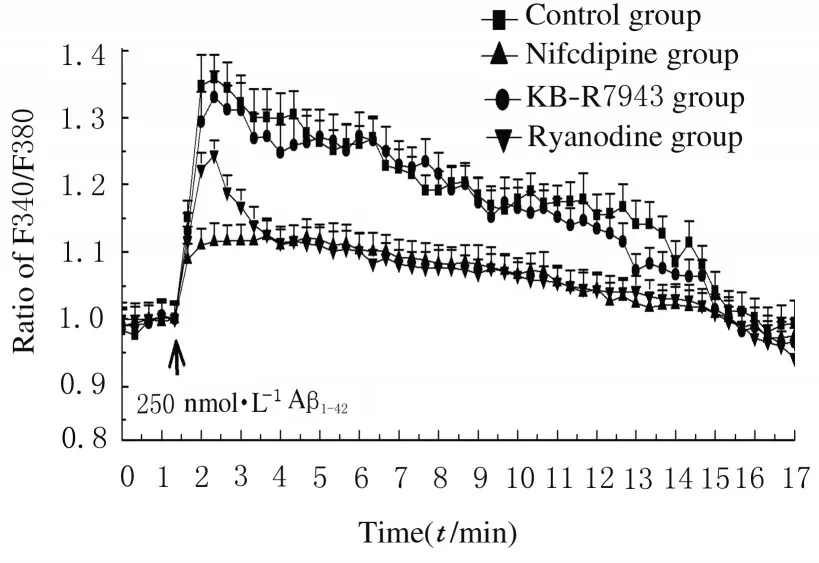
图2 荧光探针技术检测各组AS 中Ca2+水平Fig.2 Levels of Ca2+ in AS in various groups detected by fluorescence probe technique
2.2 加入不同剂量氯化锂后各组AS 中Ca2+水平取Aβ1-42刺激后3、5、7 和9 min 时间点数据进行分析(图3),与对照组比较,Aβ1-42+不同剂量氯化锂组AS 中Ca2+水平明显降低(P<0.05),Aβ1-42+不同剂量氯化锂组间比较差异无统计学意义(P>0.05)。
2.3 各组AS 的Ca2+再充功能Thapsigargin 是细胞内质网上Ca2+-ATP 酶的抑制剂,可耗竭AS 内储存的Ca2+。AS 加入Thapsigargin 作用10 min,充分耗竭细胞内储存的Ca2+,在重新加入终浓度2 mmol·L-1CaCl2后,观察Ca2+再充功能(图4)。以加入CaCl2后的2 min 内的F340/F380 值(代表Ca2+再充水平) 进行分析:与对照组(1.95±0.01) 比 较,Aβ1-42+0.50 mmol·L-1氯 化 锂 组F340/F380 值 (1.47±0.02) 明 显 降 低 (P<0.01),表明氯化锂作用2 周能明显抑制AS 内Ca2+再充功能。
2.4 各组AS 谷氨酸释放量HPLC 结果显示:与对照组比较,Aβ1-42组谷氨酸释放量明显增加(P<0.01);与Aβ1-42组比较,Aβ1-42+不同剂量氯化锂组AS 的谷氨酸释放量明显降低(P<0.01),Aβ1-42+不同剂量氯化锂组间比较差异无统计学意义(P>0.05)。见表1。
2.5 各组AS 中钙离子通道蛋白表达水平与对照组比较,Aβ1-42+ 不同剂量氯化锂组AS 中TRPC1 蛋白表达水平明显降低(P<0.05),NCX1 和CAV1.2 蛋白表达水平差异无统计学意义(P>0.05)。见图5 和表2。
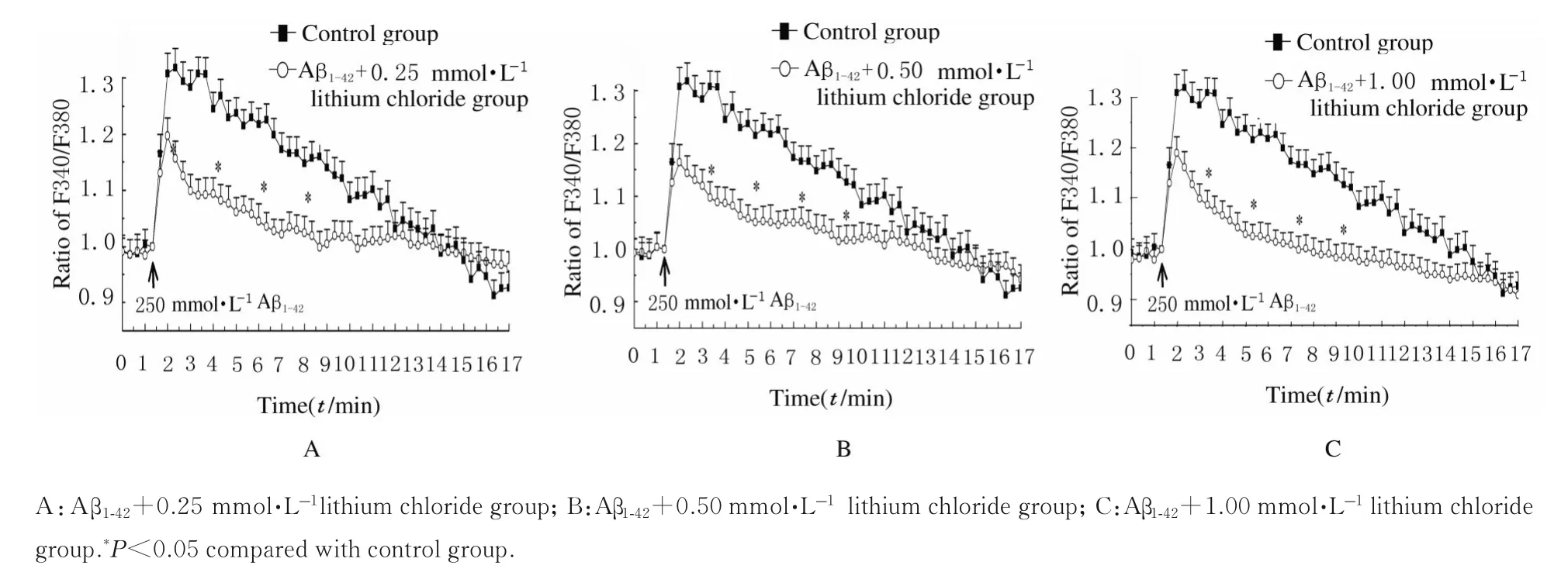
图3 加入氯化锂后各组AS 中Ca2+水平Fig.3 Levels of Ca2+ in AS in various groups after added with lithium chloride
2.6 各组海马神经元树突形态表现培养7 d 的海马神经元加入Aβ1-42刺激的ACM 作用24 h 后,Aβ1-42组海马神经元突起的生长发育被明显抑制(图6)。与对照组比较,Aβ1-42组海马神经元TDBL、PDN 和MBO 明显减少(P<0.01); 与Aβ1-42组比较,Aβ1-42+不同剂量氯化锂组海马神经元TDBL、PDN 和MBO 明 显 增 加(P<0.05 或P<0.01)。见表3。
2.7 各组海马神经元NO 释放量与对照组比较,加入Aβ1-42处理的ACM 24 h 后,Aβ1-42组海马神经元NO 释放量明显增加(P<0.01);与Aβ1-42组比较,Aβ1-42+不同剂量氯化锂组海马神经元NO 释放量明显减少(P<0.05)。加入Aβ1-42处理的不含AS的培养基24 h 后,各组海马神经元NO 释放量比较差异均无统计学意义(P>0.05)。见表4。
3 讨 论
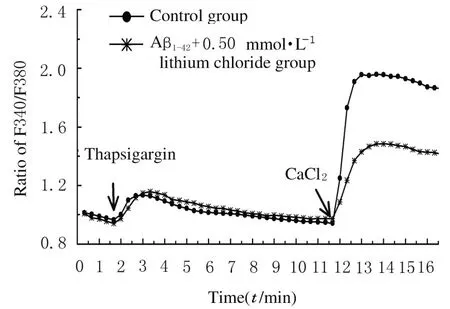
图4 荧光探针技术检测AS 的Ca2+再充功能Fig.4 Ca2+ influx of AS detected by fluorescence probe technique
表1 各组AS 谷氨酸释放量Tab. 1 Amounts of glutamate release in AS in various groups

表1 各组AS 谷氨酸释放量Tab. 1 Amounts of glutamate release in AS in various groups
*P<0.01 compared with control group;△P<0.01 compared with Aβ1-42 group.
Group Control Aβ1-42 Aβ1-42+0.25 mmol·L-1 lithium chloride Aβ1-42+0.50 mmol·L-1 lithium chloride Aβ1-42+1.00 mmol·L-1 lithium chloride Amount of glutamate release 2.08±0.24 9.46±1.39*5.29±1.03△4.69±0.78△4.14±0.83△
Aβ 沉积和细胞内神经纤维缠结是AD 典型的病理改变,然而近年来围绕抑制或清除Aβ 所研发的药物均告失败[15],提示AD 早期可溶性Aβ 引发的炎症反应或兴奋性损伤,可能是导致神经元突触结 构 和 功 能 改 变 的 主 要 原 因[2,15]。研 究[16]证 实:AD 患者脑脊液中谷氨酸表达水平异常升高,但其具体机制尚不清楚。AS 是脑内谷氨酸代谢的关键场所[4],本研究通过离体实验证明:Aβ 可促进AS释放谷氨酸,与近年来发现的Aβ 与谷氨酸兴奋性损伤相关的报道[1,17-18]一致。锂盐对AD 患者海马神经元具有保护作用,临床随机双盲试验[19]显示:长期服用小剂量锂可延缓AD 患者认知功能障碍 的 进 展。最 近 的 研 究[11,20]表 明:AS 是 锂 盐 的主要作用靶点。本研究首次证明了氯化锂对Aβ 引起的AS 谷氨酸释放具有抑制作用。有研究[5,18]表明:细胞外过多的谷氨酸引起突触外N-甲基-D-天冬氨 酸(N-nethyl-D-aspartic acid,NMDA) 受体(extrasynaptic NMDA receptors,eNMDAR)的活化是AD 最早期的病理改变,也是引起突触损伤的主要因素。因此,本研究结果为锂盐在AD 早期治疗的作用提供了理论依据。

图5 Western blotting 法检测各组AS 中TRPC1(A)、NCX1(B)和Cav1.2(C)蛋白表达电泳图Fig. 5 Electrophoregram of expressions of TRPC1(A),NCX1(B) and Cav1.2(C) proteins in AS in various groups detected by Western blotting method
表2 各组AS 中TRPC1、NCX1 和Cav1.2 蛋白表达水平Tab.2 Expression levels of TRPC1,NCX1,and Cav1.2 proteins in AS in various groups (n=3,)

表2 各组AS 中TRPC1、NCX1 和Cav1.2 蛋白表达水平Tab.2 Expression levels of TRPC1,NCX1,and Cav1.2 proteins in AS in various groups (n=3,)
*P<0.05 compared with control group.
Group Control Aβ1-42+0.25 mmol·L-1 lithium chloride Aβ1-42+0.50 mmol·L-1 lithium chloride Aβ1-42+1.00 mmol·L-1 lithium chloride Cav1.2 0.377±0.016 0.336±0.018 0.353±0.015 0.323±0.017 TRPC1 0.795±0.017 0.521±0.025*0.427±0.027*0.326±0.024*NCX1 0.831±0.024 0.806±0.033 0.836±0.029 0.826±0.028

图6 倒置相差显微镜下观察各组海马神经元树突形态表现(×400)Fig.6 Morphology of dendrite of hippocampal neurons in various groups observed under inverted phase-contrast microscope(×400)
表3 各组海马神经元TDBL、PDN 和MBOTab.3 TDBL,PDN and MBO in hippocampal neurons in various groups (n=25,)

表3 各组海马神经元TDBL、PDN 和MBOTab.3 TDBL,PDN and MBO in hippocampal neurons in various groups (n=25,)
*P<0.01 compared with control group;△P<0.05,△△P<0.01 compared with Aβ1-42 group.
Group Control Aβ1-42 Aβ1-42+0.25 mmol·L-1 lithium chloride Aβ1-42+0.50 mmol·L-1 lithium chloride Aβ1-42+1.00 mmol·L-1 lithium chloride MBO 3.56±0.72 2.28±0.42*2.86±0.55△△3.06±0.65△△3.17±0.58△△TDBL(l/μm)425±24 279±18*319±23△328±22△△332±22△△PDN 2.45±0.27 1.19±0.21*1.68±0.22△1.71±0.24△△1.74±0.22△△
表4 各组海马神经元NO 释放量Tab. 4 Amounts of NO release in hippocampal neurons in various groups

表4 各组海马神经元NO 释放量Tab. 4 Amounts of NO release in hippocampal neurons in various groups
*P<0.01 compared with control group;△P<0.05 compared with Aβ1-42 group.
NO Group ACM Control Aβ1-42 Aβ1-42+0.25 mmol·L-1 lithium chloride Aβ1-42+0.50 mmol·L-1 lithium chloride Aβ1-42+1.00 mmol·L-1 lithium chloride 5.79±0.49 8.83±0.77*7.82±0.75△7.80±0.74△7.72±0.72△Culture medium 5.23±0.33 5.57±0.46 5.49±0.51 5.67±0.48 5.44±0.44
AS 在脑内的活性与细胞内Ca2+浓度有关联,研究[5,21]表明:Aβ 引起AS 谷氨酸释放具有Ca2+依赖性。本研究证实:Aβ1-42引起谷氨酸释放的同时,也导致AS 中Ca2+水平持久性升高,氯化锂在抑制Aβ1-42所致谷氨酸释放的同时,也抑制了细胞中Ca2+水平的升高。在AS 细胞膜上,Ca2+可通过Na+-Ca2+交换、电压依赖性Ca2+通道和钙池操纵性Ca2+离 子 通 道(store operated calcium channels,SOCE) 进 入 细 胞。 在AS 内 质 网 中 有IP3和Ryanodine 敏感性内Ca2+储池释放。虽然研究[22-23]表明:细胞外Ca2+内流可引起谷氨酸释放,但在应用IP3 受体和Ryanodine 受体2 种功能性Ca2+储池的拮抗剂2APB 和Ryanodine 后,谷氨酸释放可明显被抑制,表明细胞内Ca2+储池释放,是导致谷氨酸释放的主要原因,细胞外Ca2+内流只是引起短暂的细胞内Ca2+增加。瞬时受体电位通道蛋白(transient receptor potential channel,TRPC) 是SOCE 的主要组成蛋白,内质网Ca2+储池释放后可激活胞浆膜上TRPC 通道蛋白,补充胞内Ca2+,在AS 胞 浆 膜 上 主 要 表 达TRPC1 、 TRPC4 和TRPC5 3 种亚型。研究[24-25]表明:TRPC1 在谷氨酸释放过程中起着重要作用,用TRPC1 抗体孵育细胞后,细胞内Ca2+浓度以及谷氨酸的释放均被抑制。 本研究中应用Ca2+-ATP 酶抑制剂Thapsigargin 耗竭细胞内储存Ca2+后,在细胞外重新加入2 mmol ·L-1CaCl2,也仅能部分引起细胞内Ca2+的增加。同时本研究对细胞膜上Ca2+通道蛋白测定结果表明:氯化锂对细胞内Ca2+的稳定调节作用与其下调TRPC1 表达有关,而与细胞膜上Na+-Ca2+交换和电压依赖性钙通道无关,初步证明锂盐对AS 内Ca2+浓度的抑制作用及机制。
Aβ 可促进AS Ca2+依赖的谷氨酸释放,突触间隙谷氨酸持续升高可激活神经元eNMDAR,可进一步导致神经元NO 的产生、tau 蛋白的磷酸化、caspase-3 的激活及NO 的生成被认为是导致神经元兴奋性损伤的最主要因素,NO 可引起神经元线粒体裂解、 突触损伤,进而引发认知功能下降[5,26-27]。本研究结果表明:海马神经元在加入ACM 后NO 释放量明显增加,同时神经元发生损伤性改变,而加入氯化锂作用后的ACM,海马神经元NO 释放量减少,同时神经元损伤程度减轻,但与对照组比较,氯化锂并不能完全逆转神经元的损伤性改变,说明Aβ 所致谷氨酸释放只是其导致神经元损伤的机制之一。
本研究从离体水平上证明了氯化锂抑制谷氨酸释放在神经元保护中的作用,为其在AD 治疗中的应用提供了一定的理论依据,但Aβ 可通过多途径损伤神经元,而脑内谷氨酸在海马神经元和AS 之间的调节及信息传递也是一个复杂过程,因此氯化锂如何调控TRPC1 蛋白表达,且其调控作用在AD 动物模型中是否仍然具有保护作用还需进一步研究证实。
