环木菠萝烯醇阿魏酸酯分子结构性质的密度泛函理论研究
2021-01-29李怡菲覃小丽阚建全刘雄钟金锋
李怡菲,覃小丽,阚建全,刘雄,钟金锋
(西南大学 食品科学学院,重庆,400715)
γ-谷维素是由阿魏酸与三烯萜及甾醇酯化而成的一种天然营养物质[1],其广泛存在于谷类食物中,在米糠油中含量尤其丰富(1%~3%)[2]。它的主要成分为环木菠萝烯醇阿魏酸酯、2,4-亚甲基环木菠萝醇阿魏酸酯以及甾醇类阿魏酸酯,其中环木菠萝烯醇阿魏酸酯含量最高可达38.4%[2]。LIANG等[3]研究表明γ-谷维素可通过降低胆固醇治疗血脂异常,SHU等[4]报道γ-谷维素可减轻由乙酰基酚引起的肝损伤。目前对于γ-谷维素的研究主要集中于具体物质中γ-谷维素的提取以及生理活性上,而对于从分子结构层面分析其性质的研究还尚未涉及,这对于有效开发利用γ-谷维素各种生理活性,扩宽其在食品中的应用具有重要意义。
量子化学可基于量子理论,精确、静态地研究单个或少量分子的性质、特征及反应,能够考察实验无法研究的问题,从而指导实验和生产,具有绿色、省时、过程简单等优点[5]。密度泛函理论(density functional theory, DFT)是计算分子几何和能量的重要计算技术,其精度高、速度快且克服了传统的从头算法的缺陷,目前使用最为广泛[6]。ZHENG等[7]基于概念密度泛函对短叶松素及其酯衍生物抗氧化活性进行了理论研究,结果发现其分子链长与抗氧化性具有密切的联系。WANG等[8]利用密度泛函理论评估了香豆素的抗氧化活性,并进一步计算其热力学参数,探究了香豆素清除自由基、实现抗氧化的主要机理。量子化学在多酚类物质结构性质的研究上已经有了一定的应用,但是目前还未见使用该方法对γ-谷维素的结构性质进行研究的相关报道,这在一定程度上阻碍了食品工业上对γ-谷维素的深入开发与利用。
因此,本文选用γ-谷维素中主要成分环木菠萝烯醇阿魏酸酯作为研究对象,基于密度泛函理论,对其分子结构、反应活性位点(分子表面静电势、原子电荷、前线分子轨道、概念密度泛函活性指数)、分子内弱作用力、芳香性以及红外光谱进行分析计算,以期为深入探究环木菠萝烯醇阿魏酸酯的结构性质,促进其在食品中的开发与应用提供一定的理论参考。
1 材料与方法
1.1 计算方法
环木菠萝烯醇阿魏酸酯的分子结构取自PubChem数据库,利用Gaussian 09程序进行环木菠萝烯醇阿魏酸酯的结构优化以及后续理论计算,GaussView 5.0.9用于结构可视化。采用B3LYP-D3理论方法,在6-311G + (d,p) 基组水平上完成了对环木菠萝烯醇阿魏酸酯的几何结构优化和频率振动分析,得到无虚频的能量极小结构,并在此基础上进行理论红外光谱计算,对环木菠萝烯醇阿魏酸酯的分子结构进行定性分析,采用的频率校正因子为0.969。采用M062X-D3理论方法,在带弥散的MA-DEF2TZVP基组水平上对环木菠萝烯醇阿魏酸酯进行单点能计算,通过Multiwfn[9]程序,对分子表面静电势、原子电荷、前线分子轨道进行分析,并基于概念密度泛函理论对全局描述符(电负性、化学势、化学硬度、化学软度、亲电指数以及亲核指数)和局部描述符(简缩福井函数)进行分析。利用JOHNSON等[10]提出的约化密度梯度函数(reduced density gradient, RDG),进行弱相互作用力分析;为进一步表征弱相互作用的关键临界点,依据分子中的原子理论(atom in molecular, AIM)进行电子密度拓扑分析。之后,采用M062X-D3理论方法,在DEF2TZVP基组水平,基于芳香性谐振模型(aromatic resonance model, HOMA)以及适配性自然密度划分[11](adaptive natural density partioning, AdNDP)对环木菠萝烯醇阿魏酸酯进行进行芳香性分析。
1.2 实验方法
采用KBr(光谱纯,购自天津科密欧试剂公司)压片法进行傅里叶红外光谱分析。将环木菠萝烯醇阿魏酸酯(纯度为98%,购自大连美仑生物技术有限公司)与在100 ℃下烘干后的KBr按质量比1∶100混合后,快速研磨均匀。取适量混合样品粉末压片,使用红外光谱仪(Spectrum100, PerkinElmer, Waltham, MA, USA)进行扫描,扫描范围为4 000~500 cm-1,扫描次数为32。
2 结果与分析
2.1 环木菠萝烯醇阿魏酸酯分子结构及活性位点预测
2.1.1 分子结构
在B3LYP-D3/6-311G + (d,p) 水平下优化得到环木菠萝烯醇阿魏酸酯分子几何构型如图1-C所示,频率分析无虚频,表明优化后的结构是全局能量最小值的稳定构型。优化后的结构用于后续单点能计算以及分析。

A-环木菠萝烯醇阿魏酸酯的分子式;B-优化前的分子几何构型;C-优化后的分子几何构型
2.1.2 活性位点预测
2.1.2.1 分子表面静电势
图2为环木菠萝烯醇阿魏酸酯分子表面静电势(electrostatic surface potential, ESP)分布图,其可用于预测反应活性位点,分子表面静电势越负的位置对应的原子越容易发生亲电反应,越正的位置对应的原子越容易发生亲核反应[12]。分子表面静电势图上颜色深浅反映静电势的强弱,静电势为正的区域用红色表示,静电势为负的区域用蓝色表示。其中,黄球对应静电势极大点,青球对应静电势极小点。图2显示,分子表面静电势在全局的最小值为-37.910 kcal/mol,位于酯基氧O2(图1)附近,其次为甲基氧O3、酚羟基氧O4附近(-36.400 kcal/mol),这是因为O的电负性比较强,周围电子较多密度较大,具有亲电活性,易发生亲电反应。分子表面静电势在全局的最大值(+53.850 kcal/mol)出现在酚羟基氢H99附近,表明此区域亲核性较高,容易发生亲核反应。于建成[13]对茶多酚的分子表面静电势测定结果表明,酚羟基氢附近分布着静电势极大值点(+64.210 kcal/mol),这与我们结果相似。STRUIJ等[14]实验结果发现,谷维素的抗氧化能力主要来源于阿魏酸分子的酚羟基基团,其可阻止自由基链的传递。分子静电势结果表明环木菠萝烯醇阿魏酸酯酚羟基氢具有较强的亲核反应活性,这可能与其抗氧化反应存在着一定的联系。

图2 环木菠萝烯醇阿魏酸酯分子表面静电势分布图
2.1.2.2 原子电荷
原子偶极矩校正的Hirshfeld电荷(ADCH)可表征分子内不同原子所带电荷大小,其与分子体系的亲电、亲核活性具有一定关系,其中带负电荷越多的原子相应的亲电活性可能也越强[15]。计算得到环木菠萝烯醇阿魏酸酯碳氧原子的ADCH电荷如表1所示,其中酚羟基氧O4带有最多的负电荷,酯基氧O2次之,但两者的原子电荷相差极小,表明这2个位置都具有较好的亲电活性,这与分子静电势结果酯基O2与酚羟基O4附近具有较强的亲电活性一致。这与常瑞等[16]的研究结果,N-羟乙酰神经氨酸分子的羟基氧具有多的负电荷分布,较强的亲电活性结果相似。酚羟基氢H99带有较高正电荷,表明此处亲核活性较高,易发生亲核反应。高子飞[17]研究结果表明绿原酸分子的酚羟基氢原子所带正电荷较大,这与我们的结论相似。

表1 环木菠萝烯醇阿魏酸酯碳氧原子ADCH电荷
2.1.2.3 前线分子轨道
根据前线轨道分子理论[18]可知,前线分子轨道在分子反应过程中具有重要作用。前线轨道包括最高占据轨道(highest occupied molecular orbital,HOMO)以及最低未占轨道(lowest unoccupied molecular orbital,LUMO),其中HOMO束缚电子能力较差,具有给电子性质。前线轨道能级差越小,表示分子中电子越容易发生跃迁,反应活性就越强。通过计算得到环木菠萝烯醇阿魏酸酯的前线轨道结构图(图3),其中蓝色代表轨道波函数负相位,绿色代表轨道波函数正相位。由图3可以看出,环木菠萝烯醇阿魏酸酯HOMO-LUMO能隙为7.502 eV,其中HOMO轨道都位于酚羟基氧O4、甲基氧O3、酯基O2以及苯环上酚羟基的邻位、对位碳附近,表明这些位置易受亲电试剂攻击,发生亲电反应。此结果与分子表面静电势以及原子电荷结果一致。研究表明,酚羟基具有定位活化作用,使得处于苯环上的酚羟基对位、邻位的碳原子化学性质更为活泼,易发生反应[19],这与我们的结论相似。而LUMO轨道主要分布在酯基氧(O1、O2)、双键碳(C35、C33)及酚羟基附近,表明这些位置易受亲核试剂攻击,发生亲核反应。前线分子轨道分析结果表明,环木菠萝烯醇阿魏酸酯分子结构中的阿魏酸基团上的原子是主要的亲电或亲核反应位点。

图3 环木菠萝烯醇阿魏酸酯的HOMO和LUMO轨道分布图
2.1.2.4 简缩福井函数
简缩福井函数可定量分析分子中原子的亲电亲核反应活性,其中f+表示亲核活性,f-表示亲电活性,一个分子的某原子对应的福井函数值越大,其越可能是相应类型反应的活性位点[20]。由表2可知,环木菠萝烯醇阿魏酸酯分子中,双键C33原子、酚羟基O4原子的f-值均较大,为0.083、0.078,其次为酚羟基对位C43原子,表明这些点易发生亲电反应。双键碳原子C35对应的f+值最大,为0.102,其次为C33(0.095),表明这些原子可能是亲核反应活性位点。简缩福井函数结果表明环木菠萝烯醇阿魏酸酯的亲电、亲核位点主要分布在阿魏酸基团的原子附近,此结果与前线分子轨道分析结果一致。
全局活性参数有助于分析化合物的行为以及反应性,对于预测化学反应活性具有重要意义。表3呈现了环木菠萝烯醇阿魏酸酯的全局活性参数。其中,硬度是一个重要的稳定性指标,其值为7.502。由表3可知,在电荷转移络合物形成的过程中,环木菠萝烯醇阿魏酸酯分子可能充当的是接受电子的受体中心。
表2 环木菠萝烯醇阿魏酸酯碳氧原子简缩福井函数值

表3 环木菠萝烯醇阿魏酸酯的全局活性参数
2.2 环木菠萝烯醇阿魏酸酯分子内弱相互作用分析
RDG分析可有效直观地表征弱相互作用区域及类型[21]。相互作用的性质取决于sign(λ2)ρ的值,sign(λ2)ρ>0时,表示强的互斥作用,如环、笼中的位阻效应;sign(λ2)ρ≈0时,表示范德华相互作用;sign(λ2)ρ<0时,表示强的吸引作用,如氢键等。环木菠萝烯醇阿魏酸酯的RDG二维散点图和填色等值面图如图4-A、图4-B所示。图4中蓝色部分代表吸引作用,绿色部分代表范德华力,红色部分代表强的吸引作用。在环木菠萝烯醇阿魏酸酯分子的环状结构中间均有着明显的红色梭形区域,体现较强位阻作用,对应散点图最右边的spike;环木菠萝烯醇阿魏酸酯分子中酚羟基与甲氧基等基团间的RDG等值面既有红色,又有绿色,表明既有范德华力,也有位阻作用。图5是环木菠萝烯醇阿魏酸酯的AIM电子密度拓扑分子结果图,其中黄球代表环临界点,橘球代表键临界点。其中键临界点通常出现在有吸引力的2原子之间。结合图4-B、图5可知,环木菠萝烯醇阿魏酸酯的键临界点分布的范德华力及位阻作用,而环木菠萝烯醇阿魏酸酯的环临界点主要分布在RDG等值面的空间位阻处。以上结果表明维持环木菠萝烯醇阿魏酸酯分子内部稳定的作用力主要是范德华力及空间位阻作用力。

A-散点图;B-等值面图
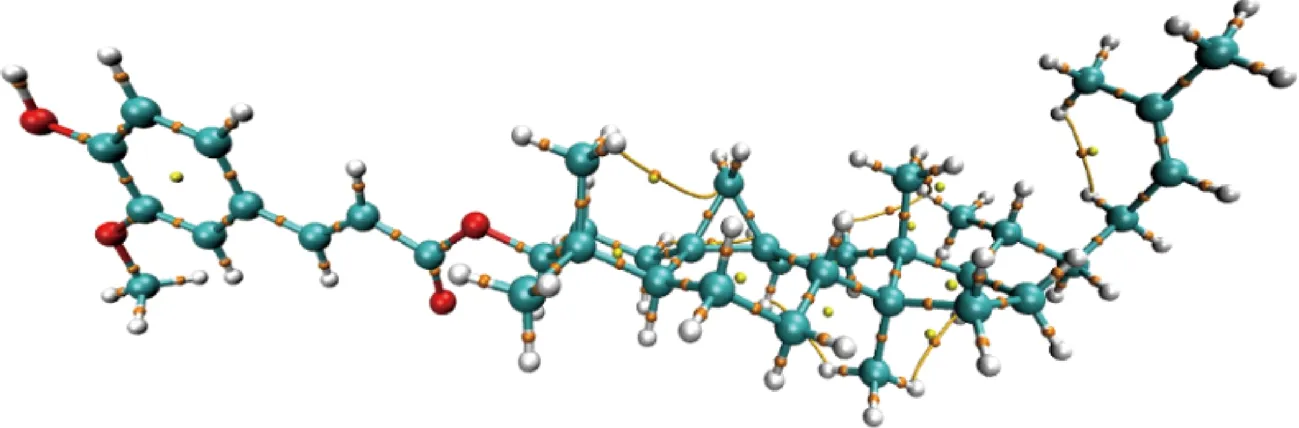
图5 环木菠萝烯醇阿魏酸酯电子密度拓扑分布
2.3 环木菠萝烯醇阿魏酸酯的芳香性
HOMA是最为常用的基于几何结构衡量芳香性的方法[22]。HOMA越接近1,说明芳香性越强;HOMA为1时,表明所有键长长度相同。计算分析环木菠萝烯醇阿魏酸酯环状结构的HOMA,结果表明环木菠萝烯醇阿魏酸酯中苯环结构的HOMA为0.960,接近1,这表明其具有较强的芳香性。采用AdNDP进一步分析环木菠萝烯醇阿魏酸酯的芳香性和成键特性[23]。由图6可知,环木菠萝烯醇阿魏酸酯结构中的具有3个6中心2电子(6c-2e)中心轨道,其形状与苯分子的占据的pi正则分子轨道相似,这表明环木菠萝烯醇阿魏酸酯分子的阿魏酸基团中构成了局部类似苯的六中心共轭体系,证明了环木菠萝烯醇阿魏酸酯具有芳香性。实验表明,谷维素具有强抗氧化性是由于其阿魏酸基团中的共轭体系形成了较为稳定的自由基,在脂质自动氧化过程中起到了终止自由基链式传递的作用[24]。芳香性分析结构证明了环木菠萝烯醇阿魏酸酯分子中共轭体系的存在,这与其抗氧化性有着一定的联系。
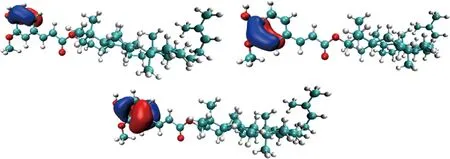
图6 环木菠萝烯醇阿魏酸酯的AdNDP键合图
2.4 环木菠萝烯醇阿魏酸酯的红外光谱


A-理论光谱图;B-实验光谱图
3 结论
本文基于密度泛函理论对环木菠萝烯醇阿魏酸酯分子结构、反应活性位点(分子表面静电势、原子电荷、前线分子轨道、概念密度泛函活性指数)、分子内弱作用力、芳香性以及红外光谱进行分析。环木菠萝烯醇阿魏酸酯酚羟基氢原子(H99)附近具有分子表面静电势极大点(53.580 kcal/mol),且H99带有较高正电荷(0.352 a.u.),表明其具有较强的亲核活性,易发生亲核反应。环木菠萝烯醇阿魏酸酯酚羟基氧原子(O4)附近具有分子表面静电势极小点(-36.400 kcal/mol),且O4具有较高负电荷(-0.378 a.u.),较高f-值(0.078),表明其具有较强的亲电活性,易发生亲电反应。环木菠萝烯醇阿魏酸酯分子的HOMO轨道都位于酚羟基氧O4、甲基氧O3以及苯环上酚羟基的邻位、对位碳附近,表明这些位置易受亲电试剂攻击,发生亲电反应。而LUMO轨道主要分布在酯基氧(O1、O2)、双键碳(C33、C35)附近,表明这些位置易受亲核试剂攻击,发生亲核反应。简缩福井函数表明酚羟基氧O4的f-值(0.078)大于其他位置氧原子,其具有较强的亲电活性;双键碳原子C33附近的f+值(0.095)较大,其具有较强亲核活性。弱相互作用分析可得环木菠萝烯醇阿魏酸酯分子结构的稳定主要与分子内范德华力及位阻作用有关。理论计算红外光谱数据与实验数据较为相似,这表明借助于密度泛函理论可较好地研究环木菠萝烯醇阿魏酸酯,这可对提取或改性的环木菠萝烯醇阿魏酸酯进行光谱预测提供一定的指导作用。本文为深入开发环木菠萝烯醇阿魏酸酯生理活性,拓宽其在食品中的应用提供了一定的理论基础。
