一维硫化钼纳米线材料第一性原理计算分析
2020-12-23史卫梅
史卫梅


摘要:将第一性原理作为基础的密度泛函理论能够探索合理新型材料,通过尺寸角度对结构进行确定,实现性能良好新型材料的设计。材料理论计算能够测试合成新材料,应用到实际工业中。目前所面临的问题就是化石燃料消耗,所以要使用清洁可持续绿色能源电化学反应能够转换化学能与电能。在电化学水解过程中要求使用催化剂使反应阻力提高,使用第一性原理计算,全面分析低维纳米材料的表面催化水解性质,实现低成本、高活性的纳米构型水解催化剂的设计。
关键词:一维硫化钼纳米线;第一性原理;密度泛函理论
新材料为促进人类社会发展主要物质力量,也是新技术开发的基础。在现代社会中,新材料技术朝着智能化、复合化、功能化的方向发展,信息功能材料、高分子材料、半导体纳米材料、生物材料等最为活跃。纳米材料具有独特的构型,在催化剂材料研究过程中广泛使用。研究纳米材料技术提高了低维新型功能材料表面活性催化活动研究频率,对于加深人们对催化反应机理的理解具有重要意义。以此,本文就对一维硫化钼纳米线材料的第一性原理计算进行分析。
1计算方法
在创建模型过程中,避免相邻的1D Mo2S5纳米线作用,设置x与y方向的晶格长度为15A。本文利用MS软件CASTP中phonopy模块进行计算,得出1D Mo2S5纳米线声子谱。利用第一性原理方法对1D Mo2S5纳米线其他性质进行测试,利用椎加平面波方法PAW与冻核近似对体系中的原子核和电子作用进行全面描述。在计算1D Mo2ss纳米线电子结构时,纯DEF计算方法会低估半导体带隙宽度,通过HSE06泛函对电子结构进行计算,接近实验值,所以电子结构性质为HSE06。将Bohn-Oppenheimer分子动力学模拟(BOMD)作为一维1D Mo2S5纳米线原子,以周期性方向使晶胞能够放大十倍,设置恒物质体系,设置步长为1fs,设置模拟步长为500步。
2结果和讨论
一维尺寸作为过渡金属硫族化合物纳米结构,材料边缘部位具备量子效应和密度局域变化,也就是通过降维手段对纳米结构进行调控,利用过渡金属硫族化合物改变电子性质。1D Mo2S5纳米线具有良好的催化作用,并且其特殊结构会提高机械应力,比热容与循环稳定效率比较高,值得深入的研究。
2.1平均结合能
对1D Mo2S5纳米线链状与管桩结构平均结合能进行研究,并且对其相对稳定性进行分析。图1为平均结合能和单元数n的关系,通过图1表示,平均结合能1NL与2NL根据单元数n不断增加而减少,增加其稳定性,两端为羟基结构相反。1NLW与3NTW并没有具备完全配位饱和的纳米结构虽然在n增加稳定性相对降低,但是还是比两端都是0的就结构稳定,表示链状终端为最强反应活性区域。3MR管状结构比较稳定,主要是由于管状结构中具备稳定硅痒四面体方式,提高了其稳定性。全部系列平均结合能都在单元数增加过程中不断收敛。
2.2能隙
为了对电子结构与反应活性进行研究,对链状与管状结构最高占据轨道与最低末占据轨道的能隙Eg进行分析。图2为结构能隙的变化关系,和平均结合能变化趋势相同,能隙在n不断增加的过程中为单调变化,而且变化幅度在不断的缩小。总而言之,完全配位饱和链状与管状结构能隙比两端为0链状能隙要大。3NTW能隙是最大的,处于8.0-8.5eV之间。另外,1NLW与3NTW的能隙具有明显的尺寸效应依赖关系。在n>14的时候,1NL与2NL能隙变化幅度在不断的增加,能隙逐渐和常数接近。两端为羟基的1NLW与3NTW能隙在计算长度范围中无法趋于长度。能隙和1/n为线性关系,拟合得出函数关系为:
R指的是相关系数,R和1越接近,表示曲线拟合效果更好。在n不断增加的过程中,1NLW能隙不断缩小,2NL相反。在纳米线长度不断增加的过程中,能隙也不会出现改变,禁带宽度比较稳定。在计算范围中能隙变化幅度没有缩小,也没有稳定禁带关系。表示长度变长之后链状结构化学反应活性不会改变,管状结构反应活性不断增强,但是还比链状结构活性要弱。
2.3力学性质
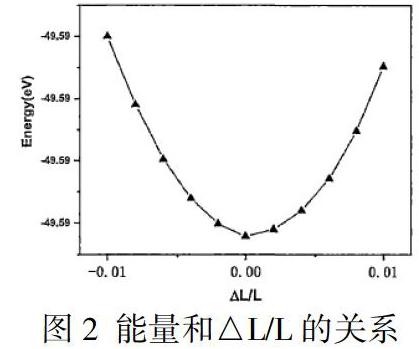
针对一维纳米线结构,机械性能尤为重要,利用以下公式对1D Mo2S5纳米线弹性模量进行计算:
公式中的C為弹性模量,E指的是体系单胞自洽能量。将z轴放方向晶格L指的是自变量,对于1D Mo2S5纳米线单胞能量作为二阶偏导数,和基态长度L0相乘,不会改变x与y中的晶格长度,z轴方向晶格长度收缩比例为±0.2%、±0.4%、±0.6%、±0.8%、±1%。得到能量和△L/L的关系,详见图2。通过DFT计算,得到1D Mo2S5纳米线弹性模量值为21.33eV/A,说明1D Mo2S5纳米线纳米线比较柔软,抵抗形变能力比较弱。
2.4电子性质
在计算1D Mo2S5纳米线电子结构时,通过PBE方法对1D Mo2S5纳米线带图进行计算,此纳米线为直接带隙半导体,宽度为0.83eV。1D Mo2S5纳米线分波态势密度图支持能带图。因为1D Mo2S5纳米线电子中的相互作用会导致出现误差,纯DFT计算电子结构会的低估体系能带宽度,通过HSE06方法能够计算1D Mo2S5纳米线纳米线带隙。对比PBE计算结果,HSE06在对1D Mo2S5纳米线进行计算的过程中会增加带隙宽度,和单层2H Mo2S5纳米线间接带隙宽度比较接近。
3结束语
通过本文研究表示,纳米材料和传统材料对比具有特殊性质,此特性包括比表面积与尺寸效应。优化材料结构,对不同结构自洽能量对比,选择额最低能量,使其成为1D Mo2S5纳米线最有构型。通过测试结果表示,1D Mo2S5纳米线较为柔软,抵抗形变能力比较弱,相对于1D Mo2S5纳米线其他催化位点和0eV更加接近,1D Mo2S5纳米线的研究在今后会备受关注,并且在HER方面的潜在应用价值不断提高。
参考文献
李筱婷. 关于一维硫化钼纳米线材料的第一性原理研究[D]. 2018.
王伟智, 徐雅飞. 一种片层堆积的一维二硫化钼纳米材料及其制备方法.
基金信息:成都职业技术学院院级科研平台,19KYPT01
