高错误率长序列的高敏感度比对
2020-12-10罗贤橦
罗贤橦,钟 诚,黎 瑶
(广西大学 计算机与电子信息学院 广西高校并行分布式计算技术重点实验室,南宁 530004)
1 引 言
以单分子实时测序技术(SMRT)[1]和Oxford Nanopore技术(ONT)[2]为代表的第三代测序平台(Third Generation Sequencing,TGS)极大地降低了测序的成本[3],可以解决人类基因组中具有复杂区域的测序问题[4].另一方面,第三代测序平台产生错误率超过15%、长度超过10k bp的长序列.测序序列长度的增加,使得一个长序列中可能包含一个完整的结构变异区域,序列比对问题从仅需要处理较短长度的错误(例如SNP错误和较短的“indel”错误),演变到需要处理较长的结构变异(转置、易位、重复和长度超过50bp的“indel”)错误,从而增加了序列比对的难度,降低了序列比对的敏感度.
针对这类错误率较高且包含错误类型较复杂的长序列比对方法有BWA-MEM[5]、BLASR[6]、Kart[7]、YAHA[8]、LAMSA[9]、Minimap2[10]和LordFAST[11]等.以BWA-MEM[5]、Kart[7]和Minimap2[10]为代表的长序列比对算法使用了双端映射(paired-end mapping,PEM)的方法,它依赖于精确匹配将种子定位到结构变异错误的任意一侧,且将种子扩展到序列全部碱基.第三代测序数据中的高错误率会导致比对算法难以找到长度较长的精确匹配,且序列长度的增长将会导致产生许多假阳性的较短的精确匹配结果,这将影响种子搜索的效率以及长序列比对的敏感度和查全率.随着第三代测序技术的发展和序列组装算法的改进[12],分割序列映射(split-read mapping,SRM)[8]成为长序列比对的一种有效策略.它更易于发现序列中的结构变异错误,且可在比对过程中减少结构变异错误对比对结果的影响.比对方法STAR[13]、YAHA[8]、LAMSA[9]、LordFAST[11]采用了该种策略.其思想是:首先将长序列分割成等长的若干片段,并将片段映射到参考基因组,生成片段定位的侯选位置,然后以片段侯选位置为依据,根据片段间可能存在的“错误”关系将片段侯选位置依次连接起来,从而获得长序列的比对结果.其中片段定位的过程将片段与参考基因比对,生成一组满足给定编辑距离的片段侯选位置.这些比对算法在处理高错误率的长序列比对时,查全率有所降低,且对错误率高的真实数据集比对的敏感度较低.
为解决现有序列比对算法对长度更长、错误率更高的第三代测序数据比对敏感度不够高的问题,本文研究使用对高编辑距离阈值更敏感的基于Hash索引的变长种子扩展方法,通过在种子验证阶段特殊处理新型错误“均聚物”[14],以提高算法对第三代测序长序列比对的查全率,在连接序列片段前,加入对片段侯选位置的质量评估,以过滤质量不高的侯选位置、提高连接效率,且在连接片段候选位置时,对不同错误类型赋予不同罚分,计算比对相似度,以减少假阳性的结果,获得高准确度和提高敏感度.
2 方 法
将高错误率长序列与参考基因组进行比对的第一步是将每条长序列划分为若干较短的片段.第二步以基于Hash索引的变长种子扩展算法定位序列片段在参考基因组中的候选位置,在种子验证过程中将连续“插入删除”相同碱基错误所带来的编辑距离设置为1,使种子扩展算法可以有效处理第三代测序长序列中的新类型错误“均聚物”[14],以使得片段定位时可提升查全率;采用全映射比对思想,寻找所有满足编辑距离阈值的片段侯选位置.第三步连接序列片段的侯选位置,对片段侯选位置进行评分,以去除质量不高的侯选位置,然后将片段连接以构建长序列比对结果的基本骨架.第四步,补全片段间的空隙,在补全过程中引入Z-drop[5]方法,以避免连接两条不相关的片段而带来的假阳性结果.本文给出的基于分割-全映射-筛选-连接-补全的高错误率长序列比对方法的执行过程如图1所示.
下面详细地阐述基于分割-全映射-筛选-连接-补全的高错误率长序列比对方法.
2.1 片段定位
已有的种子扩展方法被设计用于长度较短且错误率较低的短序列比对(short-read alignment)[15].这类种子扩展方法一般采用BWT索引结构[16]或FM索引结构[17]储存参考基因组序列信息,这两种索引结构没有存储全部的序列信息.因此,对于高编辑距离阈值的比对敏感度较低.为了提升对高编辑距离阈值的序列比对的敏感度,本文采用对编辑距离阈值更加敏感的Hash索引结构[18],通过储存参考基因组的k阶子串k-mers出现的所有位置,来实现快速搜索大量的序列[22].与BWT[16]、FM-index[17]等索引机制相比,Hash索引对高编辑距离阈值和长度更长的序列比对具有更高的敏感度,可以找到更多的符合阈值的比对结果.但随着编辑距离阈值的增大,会出现更多的假阳性的侯选位置,此外,Hash索引结构的不足是难以处理序列中存在的“插入删除(indel)”错误.为解决上述问题,本文采用变长种子选择算法[19,20]处理“插入删除(indel)”错误,且在种子验证过程中,将连续“插入删除”多个相同碱基的错误的编辑距离设置为1,以使得在种子扩展时可以处理第三代测序长序列存在的新型“均聚物”错误[14],同时采用附加k-mer算法[21]去除可能产生的假阳性错误.

图1 基于分割-全映射-筛选-连接-补全的高错误率长序列比对过程Fig.1 Process of aligning long reads with high error rate based on split-all mapping-filter-linking-completion
2.1.1 变长种子生成
与第二代测序数据主要存在“替换”错误相比,第三代测序数据中存在的错误类型还有“插入删除”错误,以往使用汉明距离确定种子候选位置的方式,并不适用于第三代测序数据.为使得比对算法既支持处理“替换”错误,又支持处理“插入删除”错误,本文采用可以处理“插入删除”错误的变长种子筛选方法[19,20],并执行附加k-mer算法[21]以去除假阳性的侯选位置.
设测序序列片段和参考基因组之间允许的编辑距离为e,根据鸽巢原理[5],则至少选取序列片段的e+1个非重叠的k-mers,才能确保至少有一个k-mer与参考基因组的精确匹配是正确的.因此,通过将一组e+1个不重叠的固定长度的k-mer与参考基因组比对,即可获得一组侯选位置集合Seed.为了使得算法可以处理“插入删除”错误,采用充分利用不重叠的k-mer之间碱基的变长种子算法,通过将固定长度k-mer进行扩展,获得一组新的变长种子侯选位置集合BSeed.在扩展时为了提高侯选位置验证阶段的效率,优先扩展出现频率较高的k-mer,以降低变长种子候选集BSeed中候选位置的数量.经过这样处理,可以最大程度地利用待比对片段上的所有碱基进行种子的生成,使得算法可以处理“插入删除”错误,提高片段比对的准确度.图2展示了一组3-mer的变长种子的生成过程实例,其中图2(a)展示找到一组不重叠的固定长度的k-mer与参考基因组进行精确匹配所获得的一组侯选位置,其中3-mer GGG在参考基因组中有5个侯选位置.图2(b)展示对固定长度的k-mer进行扩展,通过将3-mer GGG扩展为GAGGG,得到长度为5的精确匹配,获得一组假阳性较少的侯选位置.
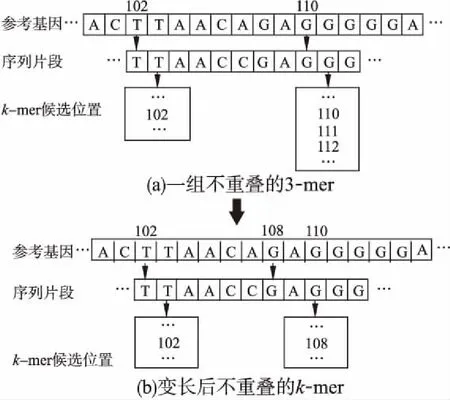
图2 变长种子生成过程实例Fig.2 Example of generating variable-length seeds
同时,为增加种子定位的准确度,采用附加k-mer的方法[21],将选取比对片段的e+2个非重叠的k-mers,作为一组候选种子,以确保其中至少有2个k-mer与参考基因组的精确匹配是正确的,进一步减少假阳性的种子侯选位置.
2.1.2 种子验证
在确定候选位置之后,对种子候选位置进行验证,仅保留所有满足编辑距离阈值小于等于e的侯选位置,作为最终的片段位置.第三代测序平台产生的测序数据中可能含有新型的 “均聚物”错误[16],即出现连续“插入或删除多个相同碱基”的错误.当遇到这种错误时,验证操作会将原本定位正确的种子过滤掉,从而降低比对的查全率.针对这种新类型的错误,本文方法在验证种子时,将“连续插入或删除同一个碱基的错误”作为一个编辑错误处理,即在验证种子阶段,当发现连续出现相同碱基的“插入/删除“错误时,则将其编辑距离定义为1,即将该种错误所造成的不匹配作为1个编辑操作,以增加片段定位的查全率,从而提升长序列比对的敏感度.
2.2 骨架构建
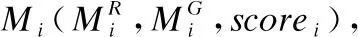
2.2.1 筛选片段侯选
在种子定位过程中,通过增大编辑距离来增加序列片段的比对侯选位置数量,这将导致假阳性侯选位置增多.因此,在骨架构建之前,对片段侯选位置进行筛选,以去除质量不高的序列片段比对的结果.
对于含有许多重复的相同碱基的序列片段,将其定位到同样具有大量重复相同碱基的参考基因组(例如人类基因组)位置上,会导致将片段定位到参考基因组中多个基因的多个不同位置上.这种片段定位得到的比对结果质量不高,会造成最终比对的假阳性结果的上升,从而影响最终比对准确度.通过统计长序列分割出的片段侯选位置,可以计算得到片段Pi的比对质量:
(1)
其中sumi为片段Pi的全部比对结果数量,sij为片段Pi在参考基因组中第j条参考基因上的比对结果数量.当Qi小于给定阈值时,舍弃该片段在第j条参考基因的比对结果.
2.2.2 片段连接
根据片段间的相互关系,将筛选后的片段进行连接,以进一步减少片段定位候选区域中的假阳性区域.从序列P0开始依次将片段的侯选位置Mi与其之前所有已连接的片段相连接,选取其中连接得分最高的连接结果,作为Mi的连接得分f(i),直至所有侯选位置均被连接到至少一条骨架上为止.本文改进后的连接得分f(i)的计算公式如下:
(2)
其中scorei为片段的侯选位置Mi与参考基因组的匹配得分,f(j)是片段Pj之前的j-1条片段的候选位置的连接得分,a(j,i)是片段Pi与它相连的片段Pj之间的关系罚分.测序序列共存在4种错误情况(如图3所示):
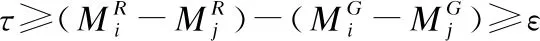
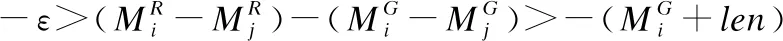
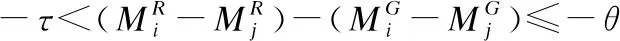
4)当Mi与Mj在参考基因组上的匹配方向相反时,片段Mi与Mj之间存在转置错误;其中ε是用于计算“插入删除”错误长度的参数,τ,θ是用于计算给定“复制”结构变异错误长度的参数[9].
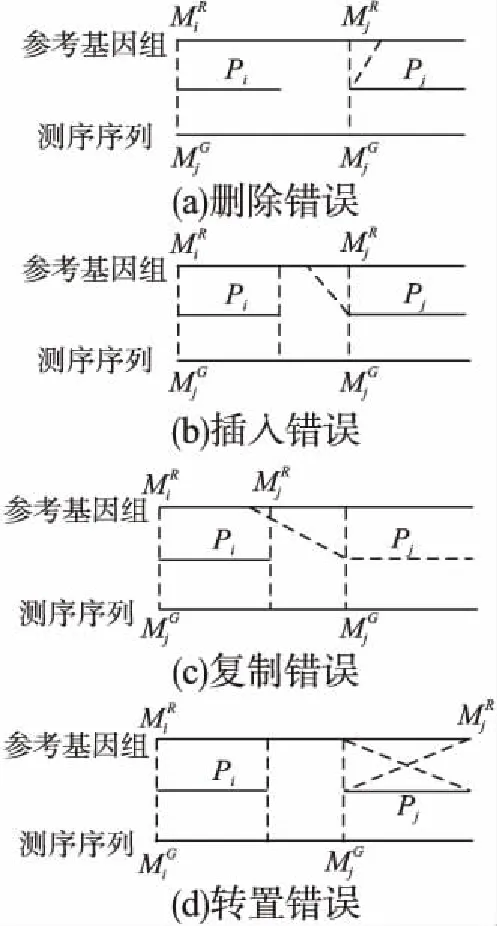
图3 测序序列的4种错误类型Fig.3 Four error types of sequencing sequences
将所有测序序列片段侯选位置连接生成一个有向无环图(DAG),从图的起点到终点的一条路径,即为一条长序列比对的连接结果,将生成的所有连接的得分进行排序,找到连接得分较大排序在前面的那些连接结果.
2.3 空隙填补
当片段连接完成后,对每条长序列的前几个连接结果执行最终比对.为了完成候选区域的碱基到碱基的对准,它以连接得分从高到低将连接好的片段之间的空隙进行匹配,匹配过程使用动态规划全局比对方法[23],将序列片段间的碱基一一对应到参考基因组侯选位置上.设定阈值,当两个片段对准得分(匹配为0,不匹配为-1)超过阈值时,则认为该条连接结果无效,这样减少将两个不相关的片段连接到一起而得到假阳性的连接结果.
长序列比对算法主要由两大部分工作组成,第一部分工作为分割序列及片段定位,通过将长序列分割为若干片段,以更快地得到较短的精确匹配的种子,对种子进行验证,得到所有满足编辑距离阈值的片段侯选位置.第二部分工作首先通过片段质量评分,去除质量不高的片段候选位置,通过片段间的连接关系去除假阳性片段侯选位置,以确保比对结果的准确度.详细比对过程如算法1所示.
算法1.基于分割-全映射-过滤-连接-补全的比对算法
输入:编辑距离阈值e,测序序列ri,i=0,1,…,rnum
输出:序列ri比对结果Si,i=,1,…,rnum
1.Begin
2.fori=1 tornumdo
3. 将序列ri以固定间距len分割为n个片段P1,…,Pn;
4.forj=1tondo
//找到所有满足编辑距离阈值e的片段候选位置选择
//Pj的e+2个非重叠k-mer作为变长种子侯选集Seed;
5.Seed[1].start←1;//种子的起始位置
6.Seed[e].end←|Pj|-1;//种子的结束位置
7.fork=1toe+2do
//将e+2个种子中出现最多的种子扩展,f为种子出现数
8.ifSeed[k].f>=Seed[k-1].fthen
9.Seed[k].start←Seed[k-1].end+1;
10.else
11.Seed[k-1].end←Seed[k].start-1;
12.endif
13.endfor
14.BSeed←Seed[1:e+2];
//将扩展后的种子Seed[1..e+1]作为新侯选集BSeed
//在Hash索引中搜索BSeed中所有种子的位置,将其存
//入片段侯选位置List[1..num]中
15.forl=1tonumdo
//验证侯选位置,筛选出符合编辑距离e的侯选位置
16.edit←0;
17.forp=1tolendo//验证
18.if片段中第p位碱基与参考基因中List[l]+p和List[l]+p-1位碱基均不同then
19.edit←edit+1;
20.endif
21.endfor
22.endfor
23. Ifedit<=ethen//编辑距离小于阈值
24.scorei←|Pj|-edit;
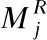
26.endif
27.endfor
28.fork=1 tondo
//统计Pk在参考基因j上的候选位置数量sij
29.sumk←0;//筛选过程sumk为片段候选结果
30.forj=1 to mdo
31.sumk←sk+sij;
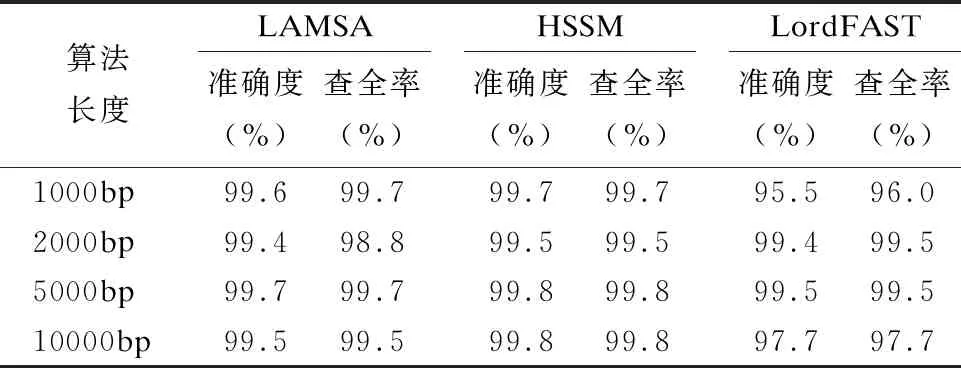
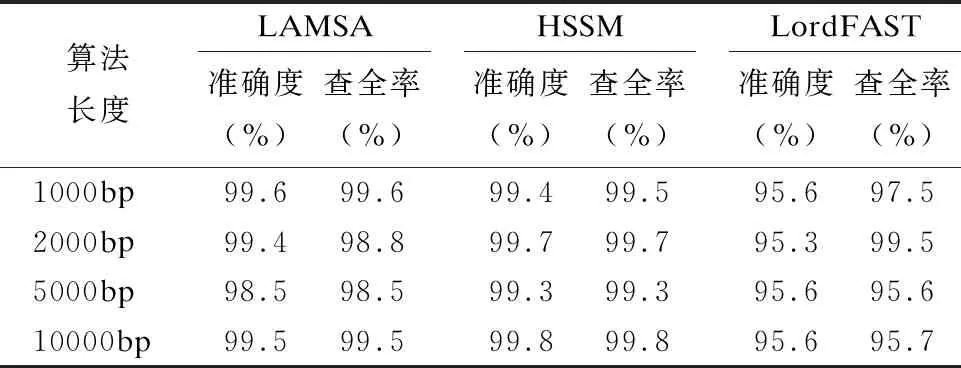
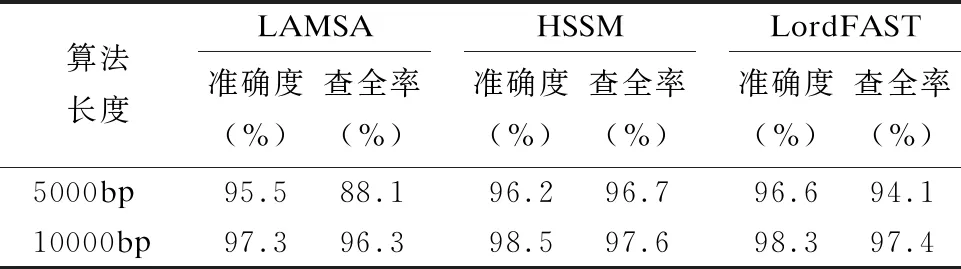
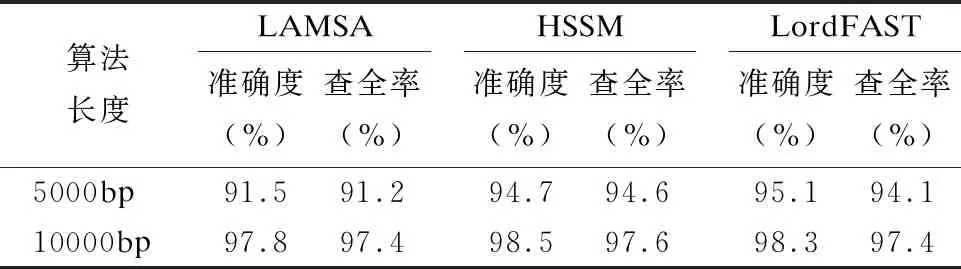
32.endfor
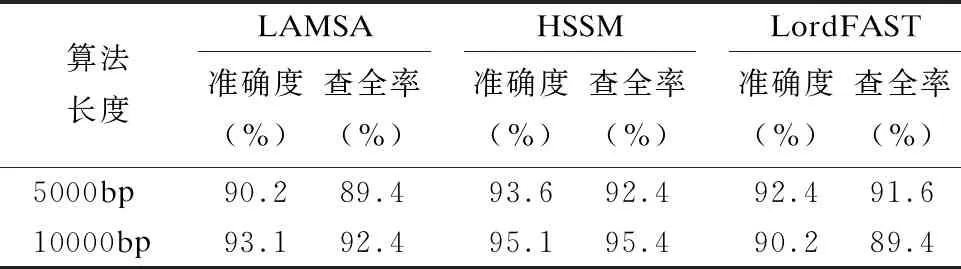
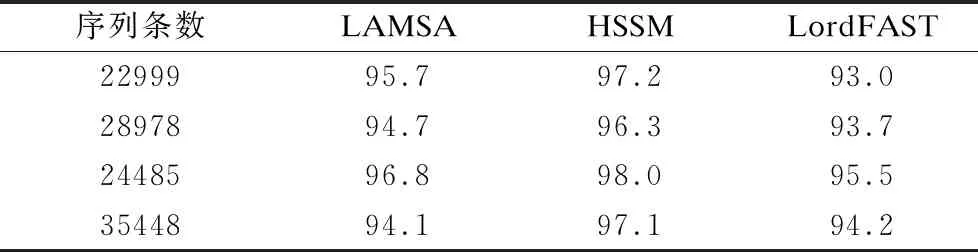
33.ifskj/sumk 34. 舍弃Pk中所有在参考基因j上的比对结果 35.endif 36.endfor 37.forl=1 tomdo //遍历所有片段,连接侯选位置Mi生成无环图 //计算候选位置Ml的最佳连接得分 39.endfor 40.根据连接得分将ri的所有连接结果进行排序; 41.选择得分最高的前几个连接结果进行片段间的空隙填补,计算序 列ri与参考基因组的匹配得分; 42.将填补后的序列匹配得分排序; 43.将最高匹配得分的结果序列在参考基因组上的起始位置,作为序 列ri最终的比对结果集合S; 44.endfor 45.end 与同类的序列比对算法LAMSA[9]和LordFAST[11]相比,本文算法采用对高编辑距离更敏感的Hash索引结构存储参考基因组,在定位序列分割片段时,通过变长种子算法[13]使得基于Hash索引的种子扩展可以处理“插入删除(indel)”错误的编辑距离,且将“连续插入/删除多个相同碱基”错误作为1个编辑距离,处理第三代测序序列特有的新型“均聚物”错误,以获得更全的片段侯选位置,确保片段定位的查全率,根据片段间的关系过滤掉片段定位侯选位置中的假阳性结果,在确保比对准确度的同时,提升查全率和敏感度. 实验使用的计算机为4核Intel(r)Xeon CPU E5-2600 V2处理器、内存容量128GB.运行操作系统Ubuntu 16.04.采用C语言编程实现算法.通过网站NCBI(1)https://www.ncbi.nlm.nih.gov/home/download/下载了公共的人类基因组数据集hg19进行比对实验. 模拟实验数据采用Wgsim(2)https://github.com/lh3/wgsim以人类基因组hg19作为参考基因组生成错误率分别为5%和10%的模拟序列数据,采用PBsim[23]以hg19作为参考基因组生成错误率分别为15%、20%和25%的含有“插入删除”和结构变异错误的长度大于等于5000bp的模拟序列. 对于生成的每条模拟序列,Wgsim和PBsim[23]都提供了模拟序列在参考基因组上的映射位置.一个模拟序列在参考基因组上的“真正”映射位置是已知的,当比对序列得出的比对位置与其模拟映射位置差值在30bp以内,则认定为该序列被比对到参考基因组中正确的位置上[7]. 序列比对准确度precision和查全率recall的计算[24]: (3) (4) 其中TP为正确映射到参考基因上的序列数量,RN为被映射到参考基因组的序列数量,N为参加映射比对的序列数量. 在模拟数据上的实验主要是为了评估序列比对算法的准确度和查全率.随着测序序列错误率的上升,比对算法采用的编辑距离也需随之增加,以获得更多的侯选位置.为此,首先对不同错误率下编辑距离阈值的选择进行实验:对10000条长度为5000bp的错误率分别为5%、10%、15%、20%和25%的模拟序列数据进行实验,测试不同的编辑距离阈值对算法的比对准确度和查全率的影响.错误率低于15%的模拟序列属于低错误率数据,选取编辑距离阈值小于5进行实验,结果如表1所示.而错误率大于等于15%的模拟序列属于高错误率数据,需要更高的编辑距离阈值,选取编辑距离阈值为5、10和15进行对比试验,结果如表2所示. 表1和表2的实验结果表明,测序序列的错误率越高,片段中所含的错误个数也就越多.此时,如果不提高编辑距离的阈值,那么将会导致丢失许多的真阳性位置,从而导致比对准确度和查全率的下降.当测序序列中错误率小于15%时,比对准确度变化不大,考虑算法处理时间,编辑距离阈值选择2即可.当错误率大于等于15%时,编辑距离阈值应选择15以获得更高的比对准确度和查全率. 表1 不同编辑距离阈值、低错误率模拟长序列比对的准确度与查全率Table 1 Accuracy and recall of simulated long-read alignment with different edit distance threshold and low error rate 表2 不同编辑距离阈值、高错误率模拟长序列比对的准确度与查全率Table 2 Accuracy and recall of simulated long-read alignment for different edit distance threshold and high error rate 下面,通过对模拟序列数据进行两组实验以评估不同序列比对算法的性能. 第一组比对实验:对长度相同、错误率不同的模拟测序序列进行实验,测试了LAMSA、LordFAST和本文算法HSSM的比对准确度和查全率.LAMSA和LordFAST算法中阈值参数选择其论文中的给定值(错误率小于15%时,容忍错误率为4%,错误率大于等于15%,容忍错误率30%),本文算法HSSM对于错误率分别为5%和10%的序列比对编辑距离阈值取值为2,对于错误率分别为15%、20%和25%的序列比对编辑距离阈值取值为15.实验结果如表3所示. 表3 不同错误率下算法比对10000条长度为5000bp的序列准确度和查全率Table 3 Accuracy and recall rate of aligning 10,000 longreads with length of 5000bp for different error rates 表3结果表明,算法HSSM采用了对高编辑距离阈值更加敏感的Hash索引方式,且在片段定位过程中针对第三代测序序列中的新错误类型“均聚物”进行处理,将所有满足编辑距离阈值的片段比对结果作为片段连接时的侯选位置,整体上获得更高的比对准确度,且获得了更多的片段候选位置,提升了定位片段的查全率,从而使得最终比对结果的查全率也随之提高. 第2组比对实验选取错误率分别为5%、10%、15%、20%和25%的10000条序列.对于错误率为5%和10%的情形,选取长度分别为1000bp、2000bp、5000bp和10000bp的4组模拟序列进行实验,实验结果见表4和表5.对于错误率为15%、20%和25%的情形,由于是模拟第三代测序的长序列数据,所以选取长度为5000bp和10000bp的模拟长序列进行对比,结果如表6、表7和表8所示. 从表4-表8的结果看,与LAMSA和LordFAST算法相比,在错误率相同且错误率较低的情形下,总体而言,本文算法HSSM的准确度和查全率有所提升.随着错误率的升高,HSSM算法获得的比对查全率高于其他两种算法.这是由于本文算法HSSM采用了基于Hash索引结构的变长种子算法,更加适应高编辑距离阈值的序列比对,可以获得更多的侯选位置,提升了片段定位的查全率,进而提升了长序列比对的查全率,且通过侯选位置的筛选和片段间可能存在的错误关系的罚分,有效去除部分假阳性错误的结果,使得长序列比对保持高的准确度.另一方面,随着模拟序列长度的增长,将长序列分割成了更多的片段,获得了更多的侯选位置,可以有效地根据片段侯选位置之间的位置关系,去除错误的比对结果,使得算法准确度随着测序序列长度的上升而提高. 表4 错误率为5%的10000条序列比对的准确度和查全率Table 4 Accuracy and recall of aligning 10,000 long readswith error rate 5% 表5 错误率为10%的10000条序列比对的准确度和查全率Table 5 Accuracy and recall of aligning 10,000 long readswith error rate 10% 表6 错误率为15%的10000条长序列比对准确度和查全率Table 6 Accuracy and recall of aligning 10,000 long readswith error rate 15% 表7 错误率为20%的10000条长序列比对准确度和查全率Table 7 Accuracy and recall of aligning 10,000 long readswith error rate 20% 表8 错误率为25%的10000条长序列比对准确度和查全率Table 8 Accuracy and recall of aligning 10,000 long readswith error rate 25% 对于真实数据集上的实验,采用比对敏感度sensitivity来评估序列比对算法[24]: (5) 其中N为匹配对准的序列条数,RN为测序数据集的序列总数. 采用PacBio测序平台产生的M130929数据集中的几组真实数据进行测试实验,结果如表9所示. 表9 真实数据集上算法的比对敏感度(%)Table 9 Sensitivity (%)of alignment algorithms on real dataset 表9的结果表明,算法HSSM使用了对高错误率更加敏感的基于hash索引结构的变长种子算法,使得hash索引可以处理“插入/删除”的错误类型,并针对第三代测序序列中新出现的“均聚物”错误类型设定其编辑距离为1进行比对,获得了更多片段候选区域,从而获得更多的比对结果,且通过采用附加k-mer算法减少了产生假阳性的比对结果,进而获得更高的比对敏感度. 综合在模拟和真实序列数据上的实验结果表明,本文算法HSSM针对第三代测序数据的特点设计改进,在获得更多侯选位置的同时,去除了假阳性的比对结果,既确保获得高的比对准确度,又获得了更高的查全率和敏感度. 通过基于Hash索引结构的变长种子算法,采用全映射思想定位长序列分割出的序列片段,可以最大程度地确保分割出的序列片段比对的查全率.分割映射的序列比对方法可以更好地处理第三代测序数据中的结构变异带来的“插入/删除”和“均聚物”错误,且通过筛选和动态连接片段侯选位置,可以得到更高的比对准确率和敏感度.高错误率的长序列比对算法中,编辑距离阈值的提高会增加种子候选数量,且随着第三代测序序列的长度越来越长,也会导致种子候选位置增加,从而使得种子验证成本增加,算法的时间开销也随之上升.第三代测序数据的高错误率的长序列比对是一个计算复杂问题.下一步工作将在借鉴全映射比对并行算法[25]的基础上,研究设计求解高错误率长序列比对问题的CPU/GPU混合并行算法,以确保比对准确度、查全率和敏感度的同时,显著加速比对完成.3 实 验
3.1 模拟数据实验


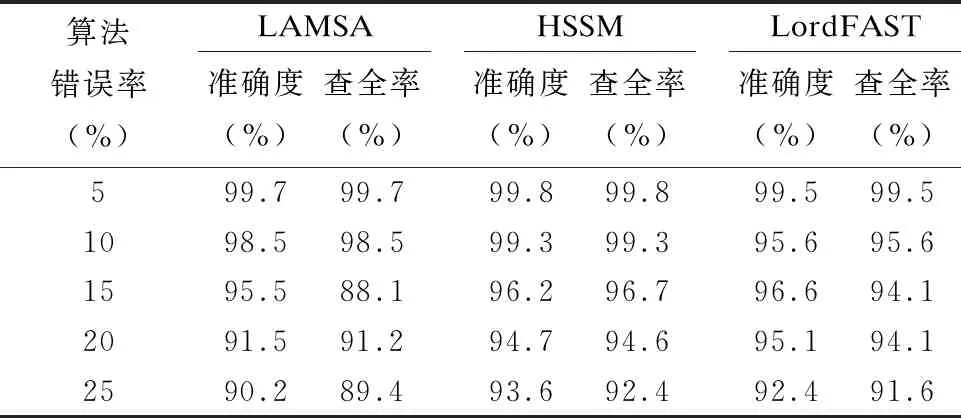
3.2 真实数据上的实验
4 总 结
