分子进化研究中多重突变的校正方法
2020-12-05李可群
李可群
(同济大学化学科学与工程学院,上海200092)
准确地估计物种分歧时间和推断其进化历史是分子进化遗传学和系统发育学的一个重要课题[1]。目前分子系统发育分析在计算物种分歧时间时大多基于分子进化速率恒定的“分子钟”假说,但绝大多数生物分子在长时间尺度和不同谱系的进化速率并不恒定,从而计算得到的结果与化石年龄往往存在较大的偏差。如计算得到的哺乳动物中真兽类的物种分歧时间平均比化石早50%~90%。基于分子数据的系统发育研究方法还不能很好地阐明哺乳动物基部问题以及哺乳动物进化过程所经历的快速辐射事件[2]。为此,本文作者提出了一种不基于进化速率恒定假说的物种分歧时间计算方法[3],并使用它发现了蛋白质和核苷酸序列分子的绝对进化速率与其进化时间之间存在定量关系,即“活化能公式”[4]。我们还计算出了“寒武纪生物大爆发”时期一些主要物种类群的物种分歧时间,所得结果与化石年龄很接近,明显好于现有基于进化速率恒定假说的分子钟方法得到的结果[5]。本文将继续对分子进化研究中两种主要多重突变类型,即回复突变和平行突变的校正方法进行探讨。
1 分子进化与泊松分布
1.1 位点突变、序列分子突变与泊松分布
泊松分布一般可用来描述稀有随机事件[6],当一个事件以固定的速率随机且独立地发生时,这个事件在某一时间段内发生的次数可用泊松分布来描述,它的概率密度函数为。若两个独立的泊松分布并列进行或先后发生,它们的平均发生次数分别为λ1和λ2,则总的随机事件仍为泊松分布,总的随机事件的平均发生次数为两个独立泊松分布平均发生次数之和,即λ=λ1+λ2,这一性质被称为泊松分布的可加性[7]。
核苷酸和蛋白质序列分子的位点突变率都很低,如蛋白质序列分子,其位点突变速率的数量级大多约为10-9位点/年[8]。一般核苷酸和蛋白质序列分子在较小的时间尺度内可视为进化速率恒定,因此在一个突变速率恒定的小时间段ta内,核苷酸和蛋白质序列分子单个位点的突变次数可认为服从泊松分布:

式(1)为单个核苷酸或蛋白质序列分子位点在该小时间段发生j次突变的概率,ka为其绝对进化速率,kata为平均突变次数,分子进化中被称之为遗传距离。特别地,单个核苷酸或蛋白质序列分子位点不发生突变的概率为p(x=0)=e-kata。对于一个有n0个被比较位点的核苷酸或蛋白质序列分子,若不考虑回复突变,有[3]

式(2)中d为核苷酸或蛋白质序列分子相对于其被比较祖先序列分子的差异位点数,k为它们的绝对进化速率,t为自其祖先序列分子而来的进化时间,p为这两个核苷酸或蛋白质序列分子的序列差异率。
1.2 分子进化与泊松分布可加性
核苷酸和蛋白质序列分子进化速率在长时间尺度内并不恒定,序列分子各位点的突变速率也存在差异[9]。为了研究方便,我们把核苷酸序列分子的进化路径拆分成由很多个足够小的时间段组成,以便在每个小时间段内核苷酸序列分子上各位点的突变速率分别恒定。同时分子进化和分子系统发育分析中一般认为生物序列分子的位点突变之间是独立的[10],这样便得到一个所谓的“泊松分布矩阵”。由于泊松分布具有可加性,“泊松分布矩阵”中同一行,即每个小时间段核苷酸序列分子各位点的泊松分布相加可得到一个新的泊松分布,新的泊松分布的遗传距离等于组成它的各位点泊松分布的遗传距离之和;而“泊松分布矩阵”中每一行相加而成的新泊松分布还可纵向逐个相加,它们的遗传距离也一样可以纵向逐个相加,后一相加关系即为文献[3]中所提及的遗传距离加和公式,或称遗传距离可加性。“泊松分布矩阵”中的每一列,即核苷酸序列分子的每个位点,它们在进化路径上各小时间段的泊松分布可以相加,后者的遗传距离亦可相应地相加。相加而成的各位点泊松分布还可相加成一个总的泊松分布,它们的遗传距离也可相加。这样,我们所研究的核苷酸序列分子在其进化路径上累积发生的突变可用该总的泊松分布来准确描述。
蛋白质序列分子的进化情形与核苷酸序列分子相似。不过两者也有不同的地方,编码序列中三个连续的核苷酸分子组成的密码子对应一个氨基酸,其中引起氨基酸变化的叫非同义密码子,不引起氨基酸变化的密码子叫同义密码子。蛋白质序列分子的“泊松分布矩阵”与核苷酸序列分子的“泊松分布矩阵”相比,缺失由同义密码子组成的那部分泊松分布(它们的遗传距离为零)。因此,对于相同的进化时间段,蛋白质序列分子的进化速率一般小于其对应的核苷酸序列分子进化速率。
2 物种分歧时间计算中回复突变和平行突变的校正方法
2.1 蛋白质序列分子中回复突变和平行突变的校正方法
物种分歧时间计算一般多使用蛋白质序列分子。我们首先讨论蛋白质序列分子中回复突变和平行突变的校正。
计算生物物种分歧时间时,因祖先蛋白质序列分子难以得到,所以一般通过比较现生同源蛋白质序列分子来计算分子绝对进化速率和物种分歧时间。文献[3]给出了图1所示的两个同源蛋白质序列分子与它们共同祖先蛋白质序列分子之间的遗传三角形关系,一个蛋白质序列分子所有已突变氨基酸位点与其同源蛋白质序列分子相同位点相比等同于与它们祖先蛋白质序列分子相同位点相比。
若遗传三角形中两个同源蛋白质序列分子的绝对进化速率分别为k1和k2,与它们共同祖先序列分子的序列差异率分别为p′1和p′2,即和。另设两个同源蛋白质序列分子自它们共同祖先序列分子的物种分歧时间为t,则根据式(2)有

图1 [3] 回复突变和平行突变校正方法的示意图

而两个同源蛋白质序列分子共同突变位点的突变概率为p′1p′2=(1-e-k1t)(1-e-k2t)。两个同源蛋白质序列分子比较时需考虑以下两种突变类型的校正,即需考虑它们对序列差异率的影响:
(1)回复突变:同源蛋白质序列分子的氨基酸位点多次突变后形成与其祖先序列分子同一位点相同的氨基酸。若一个蛋白质序列分子发生回复突变后,其同源蛋白质序列分子对应的相同位点未发生突变,则会使两者的序列差异率减少,故需进行校正。
由于存在20种基本氨基酸,若它们彼此突变的概率相同,两同源蛋白质序列分子的回复突变概率分别为式(3)中各自突变概率的。不难理解,两个同源蛋白质序列分子中一个蛋白质序列分子发生回复突变,而另一个蛋白质序列分子相同位点未发生突变的概率分别为p1=0.05(1-e-k1t)×e-k2t,p2=0.05(1-e-k2t)×e-k1t。两个同源蛋白质序列分子相同位点同时发生回复突变的情形放在平行突变校正中考虑。
(2)平行突变:两个同源蛋白质序列分子的相同位点同时突变成同一种氨基酸。平行突变也会使两个同源蛋白质序列分子的序列差异率减少。平行突变的概率p″为
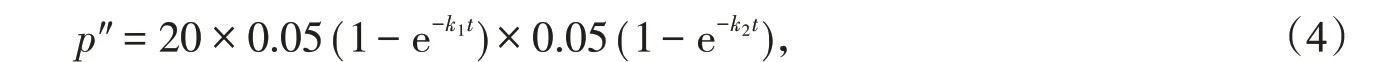
式(4)中后两个乘积项分别为两个同源蛋白质序列分子的相同位点同时突变成某一种相同氨基酸的概率,它们分别为式(3)中各自序列分子突变率的。而式(4)中第一个乘积项表示有20种可能形成的氨基酸。两个同源蛋白质序列分子的回复突变和平行突变导致两者序列差异率减少之和为
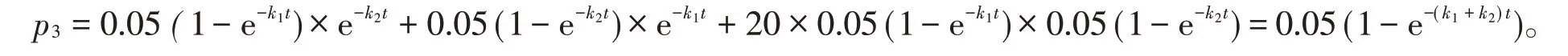
由文献[3]可知,未校正时,两个同源蛋白质序列分子的差异率为1-e-(k1+k2)t,故两个同源蛋白质序列分子在校正回复突变和平行突变后的序列差异率p为两者之差,即

可以看出,需进行校正的回复突变和平行突变概率之和为其序列差异率的。式(5)为校正回复突变和平行突变后物种分歧时间的计算公式,或多重突变校正后图1中遗传三角形的定量关系式。由文献[3]可知,在该文献表1中不引入较大相对误差的情况下,使用替代式(5)中e-(k1+k2)t,可取得较好的计算效果,即式(5)可用0.475(e-k1t+e-k2t)=0.95-p替代。
蛋白质序列决定蛋白质的结构,蛋白质结构又决定蛋白质的性能。文献[11]指出,氨基酸的亲水性和疏水性对蛋白质性能的影响很大,蛋白质序列分子突变的一般性规律是:同为亲水性或疏水性氨基酸之间突变较容易,而亲水性和疏水性氨基酸之间的突变较难发生。组成蛋白质的20种基本氨基酸中亲水氨基酸和疏水氨基酸各占约10种[11-12],因此在实际计算中发现,同源蛋白质序列分子比较时需校正的回复突变和平行突变概率之和取为其序列差异率的可取得较好效果,即可使用下列计算公式:

用式(6)和本文作者给出的计算框图成功地计算出哺乳动物[13]和原生动物[14]的物种分歧时间,结果与化石年龄相符很好,明显好于现有基于分子钟假说的计算结果。文献[15]也指出,典型的蛋白质序列分子回复突变概率为其正向突变率的。需说明的是,式(6)得到的绝对进化速率是真实值的2倍[3]。
2.2 核苷酸序列分子中回复突变和平行突变的校正方法
核苷酸序列分子中回复突变和平行突变的校正方法基本上与蛋白质序列分子相似。但核苷酸只有4种,即嘌呤核苷酸和嘧啶核苷酸各2种。核苷酸序列分子的位点突变可分为嘌呤核苷酸和嘌呤核苷酸之间,嘧啶核苷酸和嘧啶核苷酸之间的转换突变,以及嘌呤核苷酸和嘧啶核苷酸之间的颠换突变。核苷酸序列分子中转换概率一般大于颠换概率,如线粒体分子中转换与颠换概率比为15∶1或更高[16],其核苷酸转换突变中出现回复突变和平行突变的概率与颠换突变中出现回复突变和平行突变的概率比值为225∶1或更高,前者远大于后者,后者的概率可忽略。此时同源核苷酸序列分子比较时需校正回复突变和平行突变的概率之和为其序列差异率的。按照类似于蛋白质序列分子回复突变和平行突变的校正方法,可以得到核苷酸序列分子的相应校正公式为0.25(e-k1t+e-k2t)=0.5-p。由此得到的核苷酸序列分子绝对进化速率也是真实值的2倍。
由于蛋白质序列分子位点实际可发生突变的氨基酸种类约为10种,而上述研究体系中核苷酸序列分子位点实际可发生突变的核苷酸种类为2种。若10种氨基酸和2种核苷酸各自相互之间突变概率相同,根据文献[3],蛋白质和核苷酸序列分子自其祖先蛋白质或核苷酸序列分子的最大遗传距离分别约为2.3和0.7,蛋白质序列分子的最大遗传距离大于核苷酸序列分子。
3 结束语
根据泊松分布的相加性,我们提出了核苷酸和蛋白质序列分子进化的“泊松分布矩阵”模型,它能帮助我们进一步了解这些序列分子进化的机理,并可用来推导遗传距离可加性等计算公式。我们还提出了分子进化研究中回复突变和平行突变的校正方法,推导出了校正两种突变后的物种分歧时间计算公式,实际计算表明它们能得到满意的结果。在后续工作中,我们将进一步探讨文献[3]中替代公式能提高计算效果的原因和计算时物种选择规则,以期为解决目前依然争议较多的生物物种系统发育问题提供新的思路和方法。
