SOX10点突变致Waardenburg综合征2型一家系分析
2020-11-10刘薇于永波陈敏李蓓张杰
刘薇 于永波 陈敏 李蓓 张杰
国家儿童医学中心,首都医科大学附属北京儿童医院耳鼻咽喉头颈外科儿童耳鼻咽喉头颈外科疾病北京市重点实验室,100045
Waardenburg综合征(Waardenburg syndrome,WS)是一种较为常见的综合征型遗传性聋,人群发病率约为1/42000,占先天性耳聋的2%[1,2]。其病因可能是由于神经嵴细胞发育缺陷或障碍而出现细胞异常增殖、生存、迁徙和分化,而黑色素细胞、听觉神经细胞及结肠神经节细胞均源自神经嵴细胞,因此WS是一类以耳聋和着色异常为主要特征的综合征性疾病[3]。根据临床症状不同,将该病分为WS 1-4四个临床亚型[4],每一个临床亚型的意义各有差异,WS的基因突变临床上具有高度异质性,这些基因相互作用,在疾病的发生和发展中占有不同地位。WS1主要致病基因是PAX3;WS2目前已报道的致病基因包括MITF、SOX10、SNAI2;WS3致病基因为PAX3;WS4致病基因包括SOX10、EDNRB和EDN3。确定基因突变位点,从分子水平上了解疾病的病因,对于先证者自身预后及疾病的全面探究均有重大意义。本研究通过探究一WS2家系的临床表型和基因分析,为WS的临床鉴别提供了思路,同时发现了SOX10基因一点突变,该位点在Deafness Variation Database数据库中未收录,属于新发突变,具体介绍如下。
1 材料与方法
1.1 病例资料
本家系为就诊于首都医科大学附属北京儿童医院耳鼻咽喉头颈外科门诊的一山西家系。共计2代,先证者及其父母。绘制家系图谱(图1),签署知情同意书后,对先证者及其父母进行全面查体(包括毛发、皮肤色素、骨骼肌肉、四肢关节、眼科)及临床听力学评估,详细询问既往史、家族史,每人抽取5ml外周静脉血以进行基因组DNA制备及候选基因突变筛查。
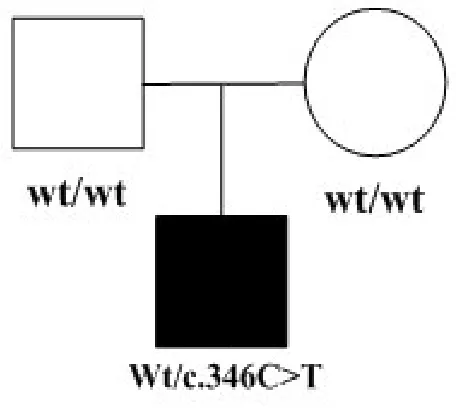
图1 家系图谱Fig.1 Family Tree
1.2 研究方法
1.2.1 听力学检查
先证者行听性脑干反应阈值及潜伏期(Auditory brainstem response,ABR)、稳态听觉诱发电位(Auditory steady state potential response,ASSR)、声导抗、畸变产物耳声发射(Distortion product otoacoustic emission,DPOAE)和行为测听;先证者父母行纯音测听检查。ABR阈值≥80dBnHL者判定为极重度感音神经性聋。ABR刺激类型为click、声音强度单位为dBnHL;DPOAE为诊断型;ASSR最大刺激强度为35dBspl。
1.2.2 基因组DNA提取及文库制备
采用QIAam全血DNA提取试剂盒(Qiagen公司,德国),按其说明书提取基因组DNA。取起始量3μg DNA,采用Covaris S2超声仪(Covaris公司,美国)进行超声片段化。利用Nanodrop 2000样本定量检测仪(Thermo Fisher科技有限公司,美国)和Agilent2100生物分析仪(安捷伦科技公司,美国)进行质控,制备全基因组文库。
1.2.3 目标基因捕获及高通量测序
应用GenCap液相捕获目标基因技术(北京迈基诺公司),捕获与耳聋相关基因的编码外显子区域。利用Illumina Nextseq 500第二代测序仪对捕获到的区域进行双端测序,读长为150bp。
1.2.4 数据筛选与生物信息学分析
将目标区域测序数据去除测序数据中的接头和低质量数据。运用BWA软件比对到参考基因组上(hg19版本),对测序深度、均一性、探针特异性等数据进行统计分析。GATK软件对各个样本的比对数据进行多态性位点的检测,对单核苷酸多态性(single nucleotide polymorphisms,SNPs)和插入缺失突变(InDels)等数据进行统计和分析,筛选得到SNPs及 InDels在千人基因组、ESP6500si、Ex-AC_ALL、ExAC_EAS正常人群数据库频率小于等于 0.05,且 经 SIFT、PolyPhen2、MutationTaster、GERP++等数据库预测结果均为致病性的位点作为与疾病相关的候选位点。
1.2.5 Sanger测序验证
根据先证者筛选得到的候选变异位点,设计并合成需要测序的DNA片段引物,采用聚合酶链式反应(polymerase chain reaction,PCR)方法对先证者及其父母DNA样本进行扩增,使用ABI 3730xl测序仪(美国Applied Biosystems公司)以Sanger测序法进行测序,测序结果使用“Mutation Surveyor”软件与参考序列进行比对分析。
1.2.6 引物设计与合成
SOX10基因位于22q13.1区域,全长12244bp,共有4个外显子,mRNA转录本的长度为2885bp,针对基因突变位点区域上下游约200bp设计相应的引物进行扩增和Sanger测序验证。正向引物F416-B1_F序列AATCCACCCGAAGCTAGAGG,反向引物R416-B1_R序列GATGACAAGTTCCCCGTGTG。
2 结果
2.1 临床结果
2.1.1 先证者
7个月男性患者,生后对声音反应差,听力筛查未通过。无巨结肠病史,否认耳毒性药物服用史,否家族遗传病史。查体:双侧虹膜蓝染,无内眦外移,无鼻根宽大,肢体活动正常,毛发颜色大致正常,双侧耳廓外形正常,外耳道通畅,鼓膜完整,标志清晰。ABR提示双耳100dBnHL未引出,DPOAE未通过,双侧ASSR相关电位均无诱发波,行为测听提示双耳极重度感音神经性聋,声导抗双耳“A”型曲线。根据WS临床诊断标准,先证者诊断为WS2型。
2.1.2 先证者父母
语言发育正常,查体:虹膜颜色正常,无内眦异位,肢体活动正常,毛发颜色正常。纯音测听结果正常。家系图谱如图1所示。
2.2 基因结果
高通量测序结果显示先证者位于SOX10基因的2号外显子发生一处杂合突变,c.346C>T(p.Q116X),即编码区第346号核苷酸由胞嘧啶变异为胸腺嘧啶,造成氨基酸改变p.Q116X,即116位的谷氨酰胺转变为终止密码子,导致基因编码蛋白截断,从而丧失功能。HGMD数据库未有该位点的相关性报道,ClinVar数据库无该位点致病性分析结果;c.346C>T的 CADD预测 RawScore为 6.787656,PHRED分值为36,预测结果为“致病性”变异(图2)。
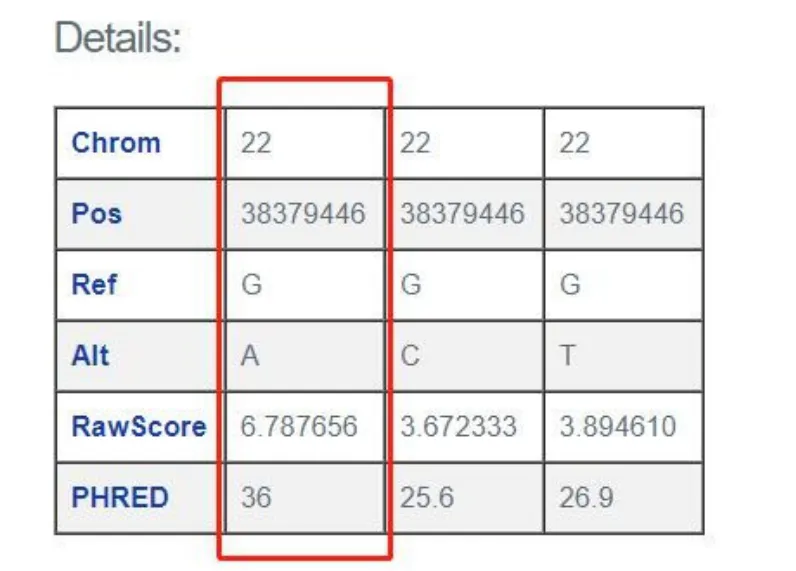
图2 突变位点CADD分值Fig.2 Score of CADD
生物信息学蛋白功能预测软件SIFT、Poly-Phen_2、MutationTaster、GERP++、REVEL分别预测分别为未知、未知、有害、有害和未知(具体见表1)。根据ACMG指南[5,6],该变异初步判定为致病性变异(Pathogenic),判定证据级别为 PVS1+PS2+PM2。经家系验证分析,先证者父母该位点均无变异,此变异为新发突变(图3)。保守性分析提示具有高度保守性,多个物种氨基酸序列一致(图4)。
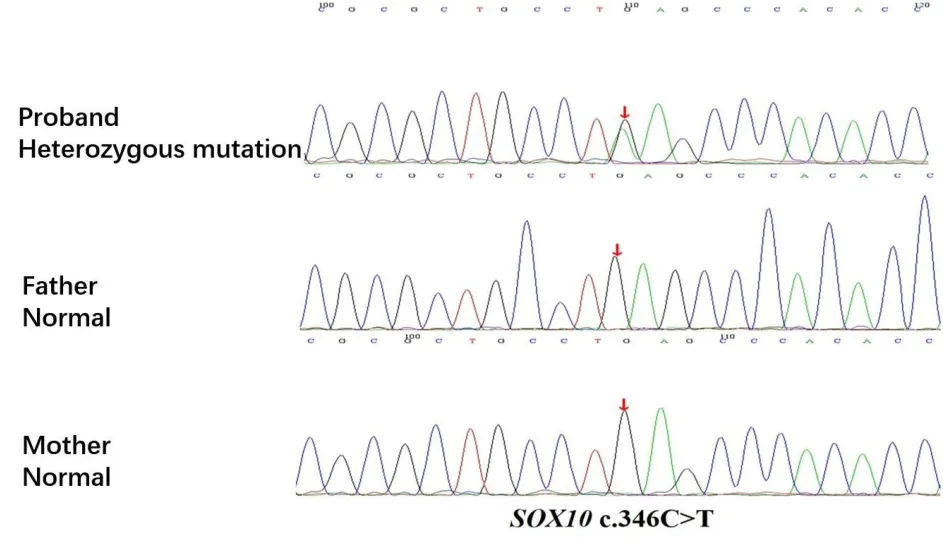
图3 SOX10基因c.346C>T突变位点Sanger测序图。红色箭头表示突变位置,先证者发生c.346C>T杂合突变,其父、其母该位置未检测到突变Fig.3 Sanger sequence of c.346C>T mutation site of SOX10.The red arrow indicates the mutation position.The proband has heterozygous mutation of c.346C>T,and no mutation is detected at the position of the patient’s father and mother
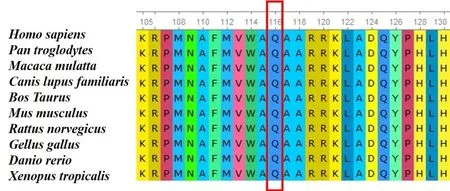
图4 p.Q116X在多个物种中具有高度的序列保守性Fig.4 p.Q116X has high sequence conservation in many species
3 讨论
WS是一类具有高度临床表型差异性及遗传异质性的疾病,即同一疾病,其临床表现可能不同,同一临床症状,其致病基因可能不同,同一致病基因的不同突变位点,其临床分型亦不尽相同,因此WS是一类值得我们深入研究以丰富人类基因突变数据库的综合征性疾病。WS的主要症状之一即感音神经性聋,从病理学上,由于耳蜗血管纹的形成需要黑色素细胞参与,黑色素细胞的缺失,导致血管纹缩小,耳蜗不发育,前庭膜塌陷及柯蒂氏器被破坏,从而导致听力损失。WS2中耳聋的发生率较高,约占50%~87%[7]。

表1 蛋白功能预测Table 1 Protein Function Predict
WS2病例中,约15%由SOX10基因突变所致[8]。SOX10基因编码的蛋白是一种转录因子,属SOX基因超级家族成员,该家族蛋白特点为含有一高度保守的高活性组分结构阈(high mobility group,HMG)。SOX10蛋白含有466个氨基酸,其主要功能是通过HMG结构域识别并结合靶基因启动子DNA,导致DNA的构象发生改变。SOX10蛋白之间可以通过单聚体或二聚体形式以多种方式激活靶基因并调节其转录,可直接或与其他转录因子协同作用于靶基因的转录,也可以和其他转录因子形成螯合复合体而间接影响靶基因的转录[8]。MITF、TYR、TYRP1、DCT、MPZ、GJB1、RET、DCT和EDNRB是其直接调控的下游靶基因[9]。多项研究表明,SOX10基因在内耳发育早期高表达,并且表达持续整个内耳发育时期,其在早期整儿耳板及耳囊广泛表达,并不仅限于内耳神经嵴来源的黑色素细胞。SOX10基因突变可导致其下游靶基因表达异常,从而造成内耳血管纹黑色素细胞和前庭及螺旋神经节胶质细胞发育异常,进而引起耳聋。
目前报道的SOX10基因突变共有61种(http://grenada.lumc.nl/LOVD2/WS/),其中导致WS2的突变7种,均为无义突变或移码突变,导致终止密码子提前出现并产生蛋白截短体而致病。本研究中,SOX10基因c.346C>T,导致氨基酸改变p.Q116X,亦为无义突变,该突变可能造成SOX10蛋白截短,c.346C>T在正常人群数据库中未出现,为低频变异,在HGMD数据库、Clinvar数据库中均未报道过该位点致病性分析的结果。既往研究表明,SOX10通过与PAX3基因相结合,进而活化下游靶基因MITF,部分学者认为,SOX10突变后完全丧失了与PAX3结合的能力[10],无法活化MITF,从而造成内耳血管纹黑色素细胞和前庭及螺旋神经节胶质细胞发育异常。课题组前期研究发现,SOX10部分结构发生变化不影响SOX10与PAX3基因的结合,但是影响MITF基因的活性,从而促使疾病的发生[11]。本研究中的SOX10突变后,可能造成蛋白截短,进而导致SOX10部分功能丧失,引起WS的发生。同样是SOX10基因突变,发生突变的位点不同,其作用机制不尽相同,引发的细胞发育异常及临床症状亦不相同,这也是这个疾病值得探索的意义所在。
本研究从临床和基因检测两方面给予患儿全面的诊断,基因诊断中发现了SOX10基因的新发突变位点,丰富了WS相关人类基因突变数据库,为我们更好的认识SOX10基因功能提供了新的线索。
