红花配方颗粒UPLC特征图谱及指标成分含量测定研究
2020-10-29李恒罗宇琴雷玉婧彭帮贵姚晓璇魏梅孙冬梅
李恒,罗宇琴,雷玉婧,彭帮贵,姚晓璇,魏梅,孙冬梅
(广东一方制药有限公司/广东省中药配方颗粒企业重点实验室,广东 佛山 528244)
红花为菊科植物红花CarthamustinctoriusL. 的干燥花,具有活血通经、散瘀止痛的功效,用于经闭、痛经、恶露不行、癥瘕痞块、胸痹心痛、瘀滞腹痛、胸胁刺痛、跌扑损伤、疮疡肿痛[1]。红花含有黄酮类、多炔类、木脂素类、生物碱类等多种化学成分[2-5],其中黄酮类成分羟基红花黄色素A和山柰素是红花中的主要活性成分[6],也是2015年版《中国药典》一部红花项下的质控指标。红花配方颗粒以符合炮制规范的红花饮片为原料,经水提、浓缩、干燥、制粒等现代工艺制成。现有的红花配方颗粒质量标准相关文献仅对部分指标的含量进行测定[7-9],未见特征图谱的相关报道。本研究采用UPLC法建立了红花配方颗粒的特征图谱,采用HPLC法建立了指标成分羟基红花黄色素A和山柰素的含量测定方法,拟为综合评价红花配方颗粒的质量提供依据。
1 仪器与材料
ACQUITY UPLC H-Class型超高效液相色谱仪,e2695型高效液相色谱仪(美国Waters公司);ME204E型电子分析天平,XP26型百万分之一电子分析天平(瑞士梅特勒-托利多公司);HWS-28型电热恒温水浴锅(上海一恒科学仪器有限公司);KQ-500DE型超声波清洗器(昆山市超声仪器有限公司);Milli-Q Direct型超纯水机(德国Merck公司)。
红花对照药材(批号:120907-200609)、羟基红花黄色素A(批号:111637-201609)及山柰素(批号:110861-201611)对照品购自中国食品药品检定研究院;乙腈、甲醇(德国Merck 公司)和磷酸(天津市科密欧化学试剂有限公司)为色谱纯,水为超纯水,其他试剂均为分析纯。15批红花配方颗粒均来自广东一方制药有限公司,编号依次为S1~S15。
2 方法与结果
2.1 特征图谱测定
2.1.1 色谱条件 色谱柱:Agilent SB C18柱(100 mm × 2.1 mm,1.8 μm);流动相:乙腈(A)-0.5%磷酸溶液(B)梯度洗脱(0~6 min,5%→8%A;6~10 min,8%→10%A;10~20 min,10%→18%A;20→28 min,18%→30%A;28~29 min,30%→90%A);柱温:30 ℃;流速:0.3 mL/min;检测波长:345 nm;进样量:1 μL。
2.1.2 对照品溶液的制备 精密称取羟基红花黄色素A对照品适量,加25%(体积分数,下同)甲醇制成含羟基红花黄色素A 195.710 2 μg/mL的溶液,0.22 μm微孔滤膜滤过,即得。
2.1.3 供试品溶液的制备 取红花配方颗粒适量,研细,精密称取0.2 g,置具塞锥形瓶中,精密加入70%(体积分数,下同)乙醇50 mL,称定质量,加热回流30 min,放冷,再称定质量,用70%乙醇补足减失的质量,摇匀,滤过,取续滤液,0.22 μm微孔滤膜滤过,即得[1]。
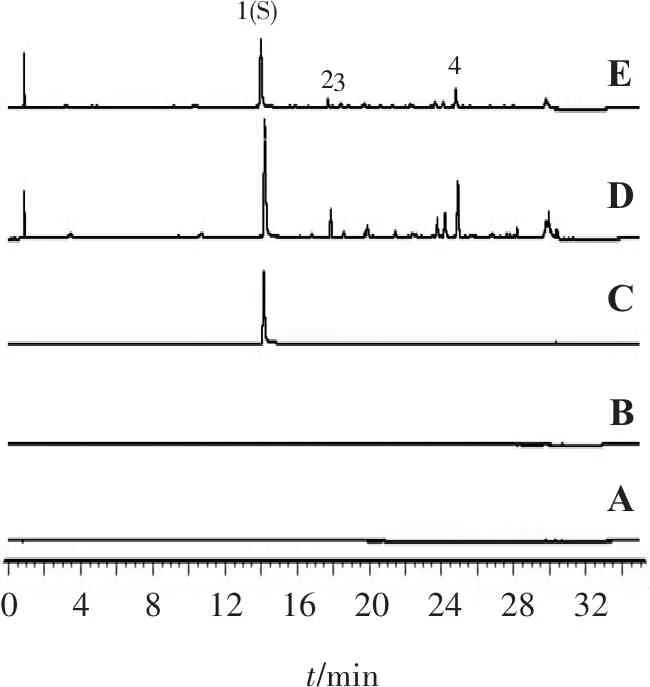
2.1.4 专属性试验 取麦芽糊精、红花对照药材及红花配方颗粒适量,按“2.1.3”项方法制备供试品溶液,按“2.1.1”项色谱条件测定,如图1。结果显示,4个共有特征峰均不受辅料、溶剂等因素的干扰,表明方法专属性良好。
2.1.5 精密度试验 取同一红花配方颗粒(编号S1)供试品溶液,按“2.1.1”项色谱条件进样6次。以1号峰羟基红花黄色素A为参照峰,计算各特征峰相对保留时间及相对峰面积的RSD分别为0.20%~0.35%,1.30%~2.08%,表明仪器精密度较好。
ABCDE1(S)234048121620242832t/min
2.1.6 重复性试验 取同一红花配方颗粒(编号S1)6份,按“2.1.3”项方法制备供试品溶液,按“2.1.1”项色谱条件测定。以羟基红花黄色素A为参照峰,计算各特征峰相对保留时间及相对峰面积的RSD分别为0.20%~0.28%,0.66%~2.68%,表明方法重复性良好。
2.1.7 稳定性试验 取同一红花配方颗粒(编号S1)供试品溶液,分别在0、2、4、6、8、12 h按“2.1.1”项色谱条件测定。以羟基红花黄色素A为参照峰,计算各特征峰相对保留时间及相对峰面积的RSD分别为0.30%~0.45%,1.21%~1.72%,表明供试品溶液在12 h内稳定。
2.1.8 特征图谱的建立 取15批红花配方颗粒,按“2.1.3”项方法制备供试品溶液,按“2.1.1”项色谱条件测定,得到15批红花配方颗粒的UPLC特征图谱叠加图见图2。可见,15批红花配方颗粒特征图谱中有4个色谱峰能稳定重现,故确定了4个特征峰,其中1号峰鉴定为羟基红花黄色素A。
2.2 羟基红花黄色素A质量分数的测定
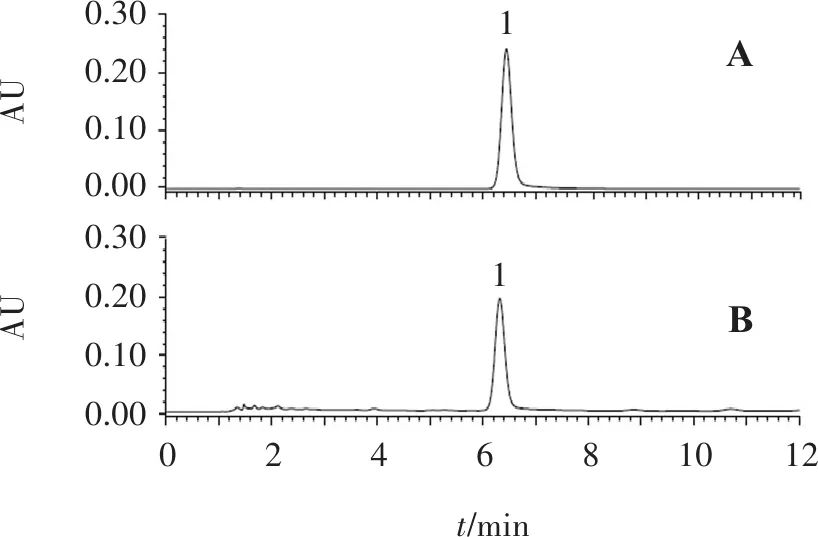
2.2.1 色谱条件 色谱柱:Agilent SB C18柱(250 mm×4.6 mm,5 μm),流动相:甲醇-乙腈-0.7%磷酸溶液(体积比26∶2∶72),柱温:30 ℃,流速:1 mL/min,检测波长:403 nm,进样量:10 μL。色谱图见图3。
2.2.2 对照品溶液的制备 精密称取羟基红花黄色素A对照品适量,加25%(体积分数,下同)甲醇制成含羟基红花黄色素A 401.235 4 μg/mL的对照品储备液。

048121620242832t/min1(S)234S15S14S13S12S11S10S9S8S7S6S5S4S3S2S1
AB110.300.200.100.000.300.200.100.00AUAU101202468t/min
2.2.3 供试品溶液的制备 取红花配方颗粒适量,研细,精密称取0.2 g,置具塞锥形瓶中,精密加入25%甲醇50 mL,称定质量,超声处理(功率300 W,频率40 kHz)40 min,放冷,再称定质量,用25%甲醇补足减失的质量,摇匀,滤过,取续滤液,0.22 μm微孔滤膜滤过,即得[1]。
2.2.4 线性关系考察 精密吸取“2.2.2”项下对照品储备液5 mL,置10 mL量瓶中,加25%甲醇定容至刻度,分别稀释成质量浓度为12.538 6、25.077 2、50.154 4、100.308 9、200.617 7 μg/mL的对照品溶液。按“2.2.1”项下色谱条件进样,测定羟基红花黄色素A的峰面积,以羟基红花黄色素A的峰面积为纵坐标、进样量为横坐标进行线性回归,得回归方程Y=3 288 646.99X+76 887.537 3(r=0.999 5),结果表明羟基红花黄色素A进样量在0.125 4~4.012 3 μg范围内与色谱峰面积呈良好线性关系。羟基红花黄色素A的检测限(S/N=3)为0.40 μg/mL,定量限(S/N=10)为1.60 μg/mL。
2.2.5 精密度试验 精密吸取“2.1.2”项下对照品溶液,按“2.2.1”项色谱条件进样6次,结果测得羟基红花黄色素A峰面积RSD值为0.15%,表明仪器精密度良好。
2.2.6 重复性试验 精密称取同一红花颗粒样品(编号S1)6份,按“2.2.3”项方法制备供试品溶液,按“2.2.1”项色谱条件测定,结果测得样品中羟基红花黄色素A质量分数RSD值为0.46%,表明方法重复性良好。
2.2.7 稳定性试验 精密吸取同一供试品溶液(编号S1),分别在0、2、4、8、12、24 h按“2.2.1”项色谱条件进样,结果测得羟基红花黄色素A峰面积RSD值0.56%,表明供试品溶液在24 h内稳定。
2.2.8 加样回收率试验 取同一批已知含量的红花配方颗粒(编号S1)9份,每份约0.1 g,精密称定,分为3组,每组分别按样品中羟基红花黄色素A质量分数的50%、100%、150%加入羟基红花黄色素A对照品,按“2.2.3”项方法制备供试品溶液,按“2.2.1”项色谱条件测定,结果测得平均回收率为96.97%,RSD为2.78%,见表1。

表1 羟基红花黄色素A加样回收率试验结果Table 1 The recovery of hydroxysafflor yellow A(n=9)
2.2.9 样品测定 取15批红花配方颗粒,按“2.2.3”项方法制备供试品溶液,按“2.2.1”项色谱条件测定,得到15批红花配方颗粒的羟基红花黄色素A的质量分数为28~39.86 mg/g,平均质量分数为33.91 mg/g;转移率为53.85%~91.36%,平均转移率为72.94%。
2.3 山柰素质量分数的测定
2.3.1 色谱条件 色谱柱:Waters X-Bridge C18(250 mm×4.6 mm,5 μm)柱,流动相:甲醇-0.4%磷酸溶液(体积比52∶48),柱温:30 ℃,流速:0.9 mL/min,检测波长:367 nm,进样量:10 μL。色谱图见图4。
2.3.2 对照品溶液的制备 精密称取山柰素对照品适量,加甲醇制成含山柰素94.163 μg/mL的对照品储备液。精密吸取上述储备液适量,加甲醇进一步稀释成质量浓度分别为38.944、4.708 15 μg/mL的对照品溶液1和2,即得。

1110121402468t/min0.300.200.100.000.300.200.100.00AUAU
2.3.3 供试品溶液的制备 取红花配方颗粒适量,研细,精密称取0.2 g,置具塞锥形瓶中,精密加入甲醇25 mL,称定质量,超声处理(功率300 W,频率40 kHz)30 min,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过,精密量取续滤液15 mL,置平底烧瓶中,加盐酸溶液(15→37)5 mL,摇匀,置水浴中加热水解30 min,立即冷却,转移至25 mL量瓶中,用甲醇稀释至刻度,摇匀,滤过,取续滤液,0.22 μm微孔滤膜滤过,即得[1]。
2.3.4 线性关系考察 精密吸取“2.3.2”项对照品溶液1各0.2、0.3、0.5、0.75、1.5、3.0 mL,分别置于5 mL量瓶中,加甲醇定容至刻度,分别配制成质量浓度为1.557 8、2.336 7、3.894 5、5.841 7、11.683 5、23.366 9 μg/mL的对照品溶液。按“2.3.1”项色谱条件进样测定,以山柰素峰面积为纵坐标、进样量为横坐标进行线性回归,得回归方程Y=4 584 822.54X-12 064.606 6(r=1.000 0),结果表明山柰素进样量在0.015 6~0.399 4 μg范围内与色谱峰面积呈良好线性关系。山柰素的检测限(S/N=3)和定量限(S/N=10)分别为0.08、0.25 μg/mL。
2.3.5 精密度试验 精密吸取“2.3.2”项对照品溶液2,按“2.3.1”项色谱条件进样6次,结果测得山柰素峰面积RSD值为0.47%,表明仪器精密度良好。
2.3.6 重复性试验 精密称取同一红花配方颗粒(编号S1)6份,按“2.3.3”项方法制备供试品溶液,按“2.3.1”项色谱条件测定,结果测得样品中山柰素质量分数RSD值为3.86%,表明方法重复性良好。
2.3.7 稳定性试验 精密吸取同一红花配方颗粒(编号S1)供试品溶液,分别在0、2、4、8、12、24 h按“2.3.1”项色谱条件进样测定,结果测得山柰素峰面积RSD值为0.61%,表明供试品溶液在24 h内稳定。
2.3.8 加样回收率试验 取同一批已知含量的红花配方颗粒(编号S1)9份,每份约0.1 g,精密称定,分为3组,每组分别按样品中山柰素质量分数的50%、100%、150%加入山柰素对照品,按“2.3.3”项方法制备供试品溶液,按“2.3.1”项色谱条件测定,结果测得平均回收率为103.19%,RSD为4.44%,见表2。

表2 山柰素加样回收率试验结果Table 2 The recovery of kaempferol(n=9)
2.3.9 样品测定 取15批红花配方颗粒,按“2.3.3”项方法制备供试品溶液,按“2.3.1”项色谱条件测定,得到15批红花配方颗粒的山柰素的质量分数为1.17~2.27 mg/g,平均质量分数为1.6 mg/g;转移率为37.42%~74.63%,平均转移率为55.57%。
3 讨论
供试品溶液制备方法考察中,分别对提取溶剂、提取方式、提取时间等因素进行了考察。特征图谱考察了25%甲醇、70%甲醇、70%乙醇、甲醇4种溶剂,羟基红花黄色素A含量测定考察了10%甲醇、25%甲醇、60%甲醇3种溶剂。分析比较所得图谱,特征图谱及羟基红花黄色素A含量测定分别采用70%乙醇和25%甲醇为提取溶剂时,成分提取最完全,图谱提供的信息量最大。羟基红花黄色素A、山柰素含量测定及特征图谱考察了提取方式(超声和加热回流)和提取时间,从提取效率和操作方便考虑,羟基红花黄色素A、山柰素含量测定采用超声提取40 min和30 min;特征图谱采用加热回流提取30 min。15批红花配方颗粒的羟基红花黄色素A和山柰素的质量分数分别为28~39.86 mg/g,1.17~2.27 mg/g,差异较小,与制剂用原药材质量和生产工艺稳定有关。
本研究对特征图谱色谱条件进行了优选,分别采用乙腈-0.5%甲酸、甲醇-0.5%甲酸溶液、乙腈-0.5%磷酸、甲醇-0.5%磷酸溶液为流动相系统进行考察,结果显示采用乙腈-0.5%磷酸可以使色谱峰达到良好的分离效果。对比了不同波长(220、254、275、345 nm)下各成分的吸收情况,当选取345 nm作为检测波长时,基线平稳、干扰较小,同时检测出更多特征成分,且各特征峰峰面积相差不大。考察不同柱温、流速及色谱柱对羟基红花黄色素A、山柰素含量测定及特征图谱的色谱条件耐用性的影响,结果显示柱温、流速的微小变化及不同厂家的色谱柱对指标成分含量、特征峰的相对峰面积和相对保留时间影响较小,表明方法的耐用性良好。
综上所述,本研究建立的红花配方颗粒UPLC特征图谱及含量测定方法稳定性、重复性良好,可为红花配方颗粒的质量控制和评价提供参考。
