二维Nb2SiTe4基化合物稳定性、电子结构和光学性质的第一性原理研究*
2020-10-22罗雄孟威威陈国旭佳管晓溪贾双凤郑赫2王建波
罗雄孟威威† 陈国旭佳管晓溪贾双凤郑赫2)3)‡ 王建波††
1)(武汉大学物理科学与技术学院,电子显微镜中心,人工微结构教育部重点实验室和高等研究院,武汉430072)
2)(武汉大学苏州研究院,苏州215123)
3)(武汉大学深圳研究院,深圳518057)
(2020年6月4日收到;2020年6月18日收到修改稿)
1 引 言
自石墨烯被成功剥离以来[1],二维层状材料因其新奇的物理特性及其在纳米器件中的特殊应用而被大家广泛关注.石墨烯得益于其特殊的狄拉克锥型能带结构而具有很高的载流子迁移率.然而其零带隙的特性限制了其在电子器件中的应用,如场效应晶体管[2].因此,打开石墨烯带隙或者寻找其他二维层状半导体材料一度成为研究的热点[3].前者可以依靠二维异质结的构建实现[4],后者则引导大家发现诸多具有高载流子迁移率的二维材料候选,如h-BN[5],过渡金属二硫化物[6],黑磷[7]等.其中得益于高周期的外壳层p轨道影响,二维碲化物常拥有较小的带隙值, 广泛应用于热电(如Bi2Te3[8])和红外光探测等领域(如Nb2SiTe4[9]).二维层状Nb2SiTe4具有空气中稳定、带隙窄、载流子类型可调以及对中红外光谱的响应良好等优异特性.其块材带隙约0.4 eV,室温下载流子迁移率可达~100 cm2·V–1·s–1[9].其单层材料的理论带隙值约为0.8 eV,可应用于光伏器件之中[10].此外,理论计算表明单层Nb2SiTe4具有强的各向异性和铁弹性,可应用于压敏器件之中[11].
然而,受限于材料晶格不匹配的问题,二维异质结器件的功能层界面处极易产生应力和缺陷.界面应力会导致二维材料晶格出现自适应畸变,影响原材料的电子结构等[12];缺陷核心会形成有害深能级捕获载流子,进而影响器件的整体性能[13].为了减少二维异质结器件界面处的晶格失配,基于同族元素的合金化方法常被用来调控功能层的晶格参数,同时保持母相结构的优良性质.此外,应力工程[14]则被用来探究二维材料的晶格参数、电子结构和光学性质等对外应力的响应,实现可控应力下的性能调控.基于此,为了优化Nb2SiTe4异质结器件的潜在界面失配,提升器件性能表达,我们通过第一性原理计算对Nb2SiTe4体系进行探究.我们采用同族元素替换法得到27种Nb2SiTe4基化合物(A2BX4)组合.通过第一性原理能量计算,筛选出稳定的组合,计算其电子结构和光学性质,为Nb2SiTe4的实验室合金化提供选择依据.为了研究实际器件中可能存在的由晶格失配造成的面内应力影响,我们针对Nb2SiTe4基化合物进行外应力的模拟, 探究了其对电子结构的影响, 为Nb2SiTe4基微纳器件的可控制备和性能调控提供了理论指导.
2 理论计算方法
我们采用VASP软件进行Nb2SiTe4基化合物的模拟.为了简化计算, 本文采用自由单层A2BX4模型进行讨论,所用晶胞的真空层为20Å(1Å=0.1 nm). 用广义梯度近似(GGA)的Perdew-Burke-Ernzerhof[15,16]来处理电子间的交换关联作用.布里渊区的K点网格划分采用Monkhorst-Pack[17]方法.K网格选为6×5×1,截断能为500 eV,原子间的相互作用力收敛标准为0.01 eV/Å.HSE06杂化泛函[18]用来计算更准确的带隙值和能带结构.声子谱的计算采用PHONOPY[19]软件中有限位移法进行.
3 计算结果与讨论
3.1 A2BX4化合物的稳定性
以Nb2SiTe4为代表的A2BX4化合物具有单斜晶体结构,空间群为P21/c,其晶体结构如图1(a)所示,A与B位阳离子被X位阴离子夹在中间,形成三明治结构.我们采用同族元素(A=V/Nb/Ta;B=Si/Ge/Sn;X=S/Se/Te)进行替换,共有27种不同A2BX4化合物组合.为了研究其理论上的稳定性,我们计算了化学势能窗口.该判断稳定性的方法广泛应用于多种材料体系.其原理是计算实验上已合成的同元素组合的竞争相或可能分解的二次相的形成焓,若存在化学势能窗口,则该化合物理论上可以稳定存在;若不存在化学势能窗口,则该化合物一定存在一条分解路径,分解为其竞争相或二次相及其组合.我们以Nb2SiTe4为例,详细介绍如何计算其化学势能窗口.首先查询MaterialsProject[20]数据库可知Nb2SiTe4存在Nb4-SiTe4和Nb3SiTe6两种竞争相,与NbTe4,Nb3Te4,NbTe2,Nb5Te4,Nb5Si3,NbSi2共有6种二次相.为了满足Nb2SiTe4的稳定条件,有如下等式:
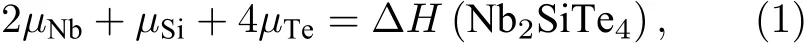
其中,µ为对应元素的化学势,∆H为对应化合物的形成焓.为了避免形成竞争相和二次相,还需满足以下关系:
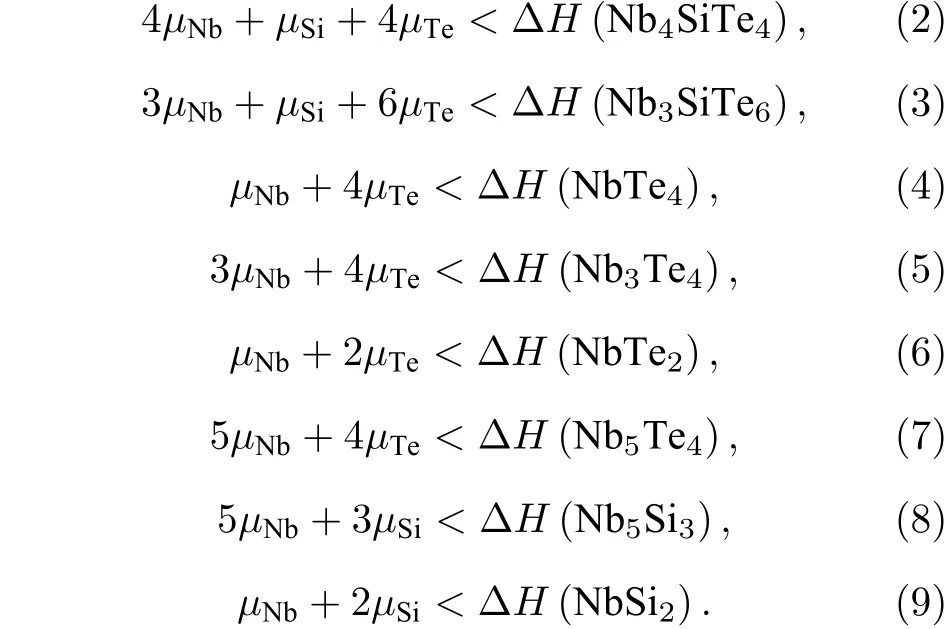

图1(a)二维A2BX4化合物的晶体结构图;(b)—(f)筛选出的5种稳定A2BX4化合物的化学势能窗口图,分别对应Nb2SiTe4,Nb2GeTe4,Nb2SiSe4,Nb2SnTe4和Ta2GeTe4.Fig.1.(a)Crystal structure of A2BX4 compounds;(b)–(f)Chemical potential windows for 5 stable A2BX4 compounds corresponding to Nb2SiTe4,Nb2GeTe4,Nb2SiSe4,Nb2SnTe4 and Ta2GeTe4,respectively.
结合不等式组(1)—(9),可以得到闭合的化学势能区间,如图1(b)阴影部分所示.阴影区域的边界即为实验室合成Nb2SiTe4时可能出现的竞争杂相,可以通过调控各组分浓度(分压)来抑制杂相的形成.为了有效合成Nb2SiTe4,应选择贫Nb富Te的环境.我们去除了不存在化学势能窗口的组合,筛选出Nb2SiSe4,Nb2SiTe4,Nb2GeTe4,Nb2SnTe4和Ta2GeTe4共5种稳定存在的A2BX4组合.其中Nb2SiTe4[21]与Nb2GeTe4[22]的多层块体材料已被实验合成,其单层材料也被理论证实可以稳定存在.Nb2SiSe4,Nb2SnTe4与Ta2GeTe43种化合物未经报道.值得一提的是,Snyde等[23]报道在实验上合成了TaGe0.6Te2,但并未给出严格的晶体结构与原子占位,不能作为Ta2GeTe4结构的参考.可以看出,A2BX4系列材料主要存在于Nb和Te组合之中.B位离子的变化对Nb-Te组合稳定性的影响相对较小.上述化合物均可以在富Se/Te环境下合成,表明阴离子的分压增大可以促进其纯相的制备.单层A2BX4的制备可以通过块材的机械剥离.Fang等[10]和Zhang等[11]结合第一性原理计算,报道了Nb2SiTe4和Nb2GeTe4的理论单层剥离能分别为0.42(0.43)J/m2和0.41(0.42)J/m2,与石墨烯,MoS2和GeS接近[11],表明其单层制备切实可行.Zhao等[9]通过胶带剥离,得到了厚度为7.5 nm(约10层)的多层Nb2SiTe4材料.对于Nb2SiSe4,Nb2SnTe4与Ta2GeTe43种未被报道的化合物,需要首先合成其块材纯相.得益于典型的三明治构型,A2BX4系列材料的层间耦合来源于上下两层邻近X阴离子的相互作用,其单层剥离能主要取决于X阴离子.当X=Te时,A2BTe4的理论剥离能接近报道的0.4 J/m2.
为了进一步确认A2BX4系列化合物的稳定性,我们通过有限位移法计算了上述3种新化合物(Nb2SiGe4,Nb2SnTe4和Ta2GeTe4)的声子谱.声子谱若存在虚频,则对应化合物的晶格存在自发沿特定方向振动的趋势,俗称“软晶格”,表明该化合物动力学不稳定,可以退化成对称性更低的结构,通过扩胞优化可以得到最终的稳定结构.如图2所示,在0 K下,3种化合物的声子谱在G-X与G-Y方向存在很小的虚频, 频率小于–0.1 THz(–3.3 cm–1).造成此种微小虚频的来源主要有两点:1)有限尺寸晶胞下对力常数的计算不够准确;2)计算中考虑单层二维材料时,丢失了第三维度的平移对称性.可以通过扩胞来消除G点处的微小虚频.考虑到扩胞成倍的增加计算成本,我们并未进行此种小虚频的完美消除.结合前文化学势能窗口的计算,足以说明Nb2SiSe4,Nb2SnTe4和Ta2GeTe43种化合物的稳定性.
3.2 5种稳定的A2BX4化合物的晶格常数,电子结构和光学性质
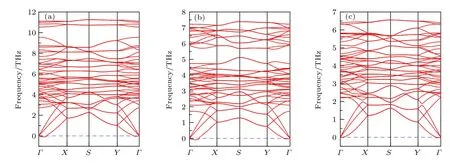
图2 0 K下基于PBE计算的声子谱(a)Nb2SiSe4;(b)Nb2SnTe4;(c)Ta2GeTe4Fig.2.PBE calculated phonon dispersions for (a)Nb2SiSe4,(b)Nb2SnTe4 and (c)Ta2GeTe4 under 0 K.
我们归纳了上述5种稳定的A2BX4单层材料的晶格常数和带隙值,如表1所示.Nb2SiSe4,Nb2-SnTe4和Ta2GeTe4均具有更窄的带隙值,其块材具有更远的红外光响应范围.为了验证计算的可靠性,我们对比了文献报道的Nb2SiTe4与Nb2GeTe4的带隙值.可以看出,基于PBE和HSE06计算的带隙值与文献报道的非常接近.Nb2SiTe4的理论晶格常数为a=6.40Å,b=7.92Å,与实验值非常吻合(a=6.30Å,b=7.90Å).通过同族元素替代,其晶格常数可以得到有效调控:a的调控范围为6.04—6.81Å,b的调控范围为7.74—8.15Å,在进行不同二维异质结构建时,有了更多的晶格选择以匹配衬底.值得一提的是Ta2GeTe4的晶格参数与Nb2SiTe4接近,却保持更窄的带隙值,可以在远红外光吸收波段替代Nb2SiTe4.
为了更精确描述A2BX4体系的电子结构,我们使用HSE06泛函计算了能带结构,并参考Nb2SiTe4进行了能带对齐.如图3所示,上述5种A2BX4材料均为间接带隙半导体,其VBM落在G点,CBM落在G-Y之间.其中:1)Nb2SiSe4带隙值(0.74 eV)比Nb2SiTe4(0.87 eV)更小,VBM位置更高.常规硫族化合物中,随着阴离子周期的增大,带隙值越来越小.这是因为其VBM通常由硫族元素的外壳层p轨道主导.随着周期的增大,p轨道的能量升高,VBM升高,带隙值减小.在A2BX4体系中,VBM来源于A位阳离子的d轨道与X位阴离子的p轨道反键作用,以d轨道主导.相较于Te,Se与Nb的反键作用更强,VBM被推的更高,更易呈现p型导电.不同于钙钛矿化合物(X'= Cl/Br/I)中,当X'由I变为Cl时,虽然VBM的s-p反键作用增强, 但是整体的VBM依然下移. 这是由于s-p反键态中p轨道主导VBM,因而p轨道能量区间决定整个体系VBM的能量位置.在A2BX4体系中,VBM(d-p反键)主要由A离子的d轨道贡献,因而X离子由Te变为Se时,其p轨道的降低不足以抵消d-p反键轨道的升高.2)当B离子从Si,Ge到Sn变化时,VBM位置逐渐升高,CBM位置变化不大.观察能带形状可以发现, VBM的带宽从1 eV (Nb2SiTe4)增大到1.5 eV(Nb2SnTe4),显示出明显的d-p反键增强效应.同时,VBM和价带电子海洋的脱离越来越远,直至形成类孤立的价带顶(Nb2SnTe4).3)对比Nb2SiTe4与Ta2GeTe4发现, 后者的VBM更高. 这是由于B位元素的变化(从Si到Ge)和A位元素变化(从Nb到Ta)的双重影响.Ta的外壳层5 d轨道在能量上高于Nb的4d轨道.

表1 A2BX4化合物的理论晶格常数与带隙值Table 1.Theoretical lattice constants and bandgaps of stable A2BX4 compounds.
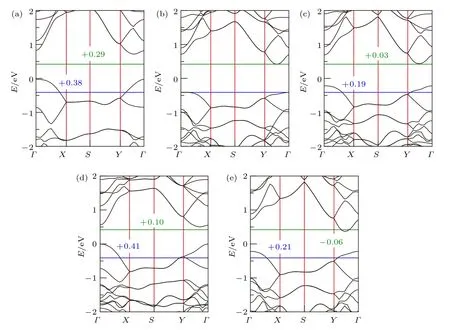
图3 HSE06计算的能带结构(a)Nb2SiSe4;(b)Nb2SiTe4;(c)Nb2GeTe4;(d)Nb2SnTe4;(e)Ta2GeTe4.其中蓝线(绿线)代表Nb2SiTe4的VBM(CBM)位置,数字表示相比Nb2SiTe4 VBM(CBM)的移动Fig.3.HSE06 calculated band structures for(a)Nb2SiSe4,(b)Nb2SiTe4,(c)Nb2GeTe4,(d)Nb2SnTe4 and(e)Ta2GeTe4,respectively.Lines represent for VBM(blue)and CBM (green)of Nb2SiTe4.Numbers indicate VBM(CBM)shifts compared with Nb2SiTe4.

图4计算的5种A2BX4光吸收谱(a)沿x方向;(b)沿y方向Fig.4.Calculated optical absorption coefficients for five A2BX4 compounds along (a)x direction and (b)y directions.
我们计算了上述5种单层A2BX4化合物的光学性质.图4(a)沿x方向,图4(b)沿y方向.阴影部分为AM1.5G太阳辐射光谱.可以看出此系列材料在红外到紫外光区间内均具有较强的光吸收特性(光吸收系数在105cm–1量级),可以应用于红外光探测和光伏器件之中.此外,A2BX4化合物沿x方向的光吸收较y方向更强,具有明显的光学各向异性,表明其在偏振光探测方面有潜在应用.
3.3 应力工程调控A2BX4化合物电子结构
外加应力可以有效调控二维材料的电子结构及相关性质,对柔性或与衬底存在晶格不匹配而产生面内应力的二维材料尤为重要.我们针对5种稳定的A2BX4单层材料,通过改变晶格参数,施加均一的双轴应力,应力大小为(a–a0)/a0,其中a0为0 K下基态的晶格常数.应力为负值时,表明施加的是双轴压缩应力,晶格常数减小;结果为正值时,施加双轴拉伸应力,晶格常数增大.如图5可以看出,外加+5%拉伸应力时,A2BX4系列化合物的带隙值均减小约0.25 eV,体系由间接带隙转变为直接带隙.外加–5%压缩应力时,A2BX4化合物中Nb2SiSe4与Nb2SnTe4的带隙值增加约0.3 eV,保持间接带隙.然而,其他3种化合物的带隙值变化异常,甚至出现减小(如Nb2SiTe4).观察压缩应力下A2BX4系列化合物的能带结构我们发现:Nb2SiTe4,Nb2GeTe4和Ta2GeTe43种化合物的VBM形状发生了变化.此种VBM和CBM的变化可以有效解释不同应力条件下化合物带隙的变化趋势问题:
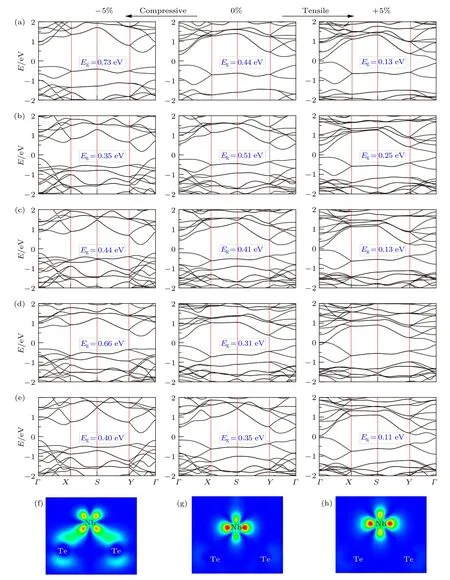
图5 PBE计算的外加±5%应力时A2BX4的能带结构图(a)Nb2SiSe4;(b)Nb2SiTe4;(c)Nb2GeTe4;(d)Nb2SnTe4;(e)Ta2GeTe4;(f),(g),(h)为对应外应力下Nb2SiTe4 VBM的电荷密度图Fig.5.PBE calculated band structures for A2BX4 under±5%strain:(a)Nb2SiSe4;(b)Nb2SiTe4;(c)Nb2GeTe4;(d)Nb2SnTe4;(e)Ta2GeTe4;(f),(g),(h)VBM charge density maps of Nb2SiTe4 under strains.
1)CBM的一致性变化.i)无应力晶格状态下,A2BX4化合物的CBM贡献主要来源于A位阳离子(Nb/Ta)的空d轨道和B位阳离子(Si/Ge/Sn)的空p轨道.X位阴离子(Se/Te)与A位阳离子形成弱的p-d反键态,与B位阳离子形成p-p反键态,此两中轨道成分共同构成CBM.ii)外加压缩应力时,A位阳离子与B位阳离子距离缩短,空d与空p轨道交叠形成A-B之间的弱p-d成键态.此弱成键态对CBM位置影响较小.同时,A/B阳离子与X位阴离子间距缩短,p/d-p轨道相互作用增强,反键态持续升高,导致CBM位置大幅升高.iii)外加拉伸应力时,A与B位阳离子间距增大,相互作用减弱,其成键态逐渐消失,对CBM影响持续减弱;A/B阳离子与X阴离子键长增大,其反键p/d-p相互作用减弱,导致CBM位置大幅度下降.由此可知,从压缩应力逐渐向拉伸应力的过渡中,CBM的主要成分不变,由A位离子空d轨道与B位离子空p轨道共同和X阴离子的反键轨道组成,该反键作用随离子间距的增大逐渐减弱,CBM位置逐渐降低.
2)VBM的反常变化.VBM反常变化的主要原因为贡献VBM的轨道特性发生了变化,我们依照图5所示能带结构,将5种A2BX4化合物分为两类:i)VBM未发生突变.有Nb2SiSe4(图5(a))与Nb2SnTe4(图5(d)).此类化合物的VBM形状保持一致,由A位阳离子的占据态d轨道主导,与少量X阴离子的p轨道形成d-p反键,如图5(g)所示.此d轨道不甚活跃,随外应力的变化,其变化不大,如图5(h)所示,VBM的位置变化也不大.此类化合物的带隙改变主要来源于对应CBM的位置改变.ii)VBM发生了突变.有Nb2SiTe4,Nb2GeTe4与Ta2GeTe4.从图5(b),(c),(e)中可以看出,施加拉伸应力时,其VBM的轨道特征并未发生改变,此时起带隙值的变化主要来源于CBM的变化.此点可以从其带隙变化均为~0.25 eV处得到证实.当施加压缩应力时,此3种化合物的VBM由原始的A离子d轨道主导,变成了A离子与X离子共同贡献.以Nb2SiTe4为例,如图5(f)所示,外加–5%应力时,其VBM在G点的主要贡献为Nb离子的d轨道与Te离子的p轨道.得益于面内晶格常数的减小,Te与周围Te离子的间距显著缩短,从0%应力下的3.9Å缩短为3.7 Å,扩宽了Te 5p轨道贡献能带的宽度,在能量上高于满d轨道,形成新的VBM.因而在压应力作用下,A2BX4化合物的带隙变化来源于CBM升高与VBM升高的竞争作用.当VBM升高更快时,体系的带隙整体呈现反常减小情况,如Nb2SiTe4.此外, VBM发生轨道反转后的能带更加分散, 其VBM的电荷密度分散在Nb-Te框架之中(如图5(f)),预测能大幅降低空穴的有效质量,促进空穴的迁移.
值得一题的是,为了节约计算成本,在讨论外应力对A2BX4体系电子结构的影响中,我们使用了PBE泛函.该泛函不能准确描述体系带隙的绝对值.然而,带隙变化的相对值却可以通过PBE泛函得到可靠的描述.类似的还有体系的能带形状,载流子有效质量等.
4 结论
通过第一性原理计算,我们系统研究了A2BX4系列化合物的稳定性,电子结构,光学性质和应力工程对其电子结构调控,确认了27种A2BX4组合中存在5种稳定结构.其中已有Nb2SiTe4与Nb2GeTe4的实验报道.Nb2SiSe4,Nb2SnTe4与Ta2GeTe4为首次报道的稳定组合,且具有类似Nb2SiTe4的强光吸收性质(a>105cm–1)和更窄的带隙值(0.51—0.74 eV).通过同族元素替换,我们可以有效调控Nb2SiTe4体系的晶格参数,实现基于A2BX4二维异质结的晶格匹配.其中Ta2GeTe4具有和Nb2SiTe4相同的晶格参数,更窄的带隙值,可在远红外光探测方面替代Nb2SiTe4.外加拉伸应力可以减小A2BX4体系带隙值.外加压缩应力时,Nb2SiSe4与Nb2SnTe4带隙增大,其他3种化合物受限于VBM的轨道反转影响,带隙值增加不明显甚至减小.该VBM轨道反转来源于Te阴离子p轨道的贡献,会降低空穴的有效质量,促进空穴的迁移.我们的结果为A2BX4二维异质结器件的设计拓宽了本体和基体的选择范围,为其器件性能的提升提供了理论指导.
