3-取代吲哚酮类抗癌药物对抗神经退行性疾病之进展
2020-10-19张在军李铭源韩怡凡
黄 凌,张在军,李铭源,韩怡凡,崔 巍
(1. 宁波市康宁医院精神科,宁波大学医学院,浙江 宁波 315020;2. 暨南大学新药研究院,广东 广州 510632;3. 澳门大学中华医药研究院,澳门 999078;4. 香港理工大学应用生物与化学技术学系,香港 999077)
随着人口老龄化的进程,神经退行性疾病已成为继心脑血管疾病、恶性肿瘤之后的第三大杀手,但是,对症治疗神经退行性疾病的药物尚未面世。目前临床使用的治疗药物虽能减轻神经退行性疾病症状,但无法阻止神经元死亡。因此,急需研发能够减慢或阻止疾病进程的神经保护药物。
最近研究发现,许多肿瘤和神经退行性疾病具有相似的特征,如都是随着年龄增长,发病率上升的疾病,最近发现胶质母细胞瘤、前列腺癌等患者罹患阿尔兹海默症(Alzheimer’s disease,AD)的比例更高,提示机体的某些信号通路可能同时对肿瘤细胞和神经元有调控作用[1]。这使人们思考抗肿瘤药物可否通过调节这些共有的信号通路阻止神经退行性疾病[2]。研究发现临床治疗慢性髓性白血病和恶性胃肠道间质肿瘤的Abl激酶抑制剂甲磺酸伊马替尼可有效抑制线粒体功能障碍,阻止神经毒性蛋白β-淀粉样蛋白(β-amyloid,Aβ)产生,因此可能“老药新用” 治疗AD[3]。
3-取代吲哚酮类化合物也兼具抗肿瘤和抗神经退行性疾病药效。3-取代吲哚酮类化合物是含有吲哚-2酮母核结构的杂环类化合物。1998年,SUGEN公司设计、合成了舒尼替尼、SU4312、SU5416等系列3-取代吲哚-2-酮类化合物。这些化合物可竞争性结合多种受体酪氨酸激酶(receptor tyrosine kinase,RTK)的ATP结合位点,进而抑制RTK的功能。由于包括血管内皮生长因子受体-2、血小板衍化生长因子受体在内的多个RTK介导了肿瘤相关信号通路,因此3-取代吲哚-2-酮类化合物可通过特异性抑制这些受体,阻止肿瘤生长和血管新生,杀死肿瘤细胞。2006年,舒尼替尼被美国食品药品管理局批准用于临床治疗肾细胞癌和胃肠间质瘤,是首个被批准用于治疗两种以上癌症的药物。SU4312、SU5416等也已进入肿瘤治疗的临床实验。合成的3-取代吲哚酮类化合物还包括SU6668、GW5074等,但这些化合物由于特异性差、副作用强等原因未能进入临床癌症治疗。
有趣的是,多种天然产物也具有3-取代吲哚-2-酮结构。上世纪七十年代,中国医学科学院血液学研究所从青黛中提取了3-取代吲哚酮类天然产物——靛玉红。靛玉红有很强的新生血管抑制活性,能抑制肿瘤生长,现已被中国药监局批准用于治疗慢性粒细胞白血病。近年来,许多靛玉红衍生物如靛玉红-3-肟、7-溴-靛玉红-3-肟也显示了良好的抗肿瘤效果,进入临床或临床前实验(Fig 1)。本文尝试从机制上探讨3-取代吲哚酮类化合物对抗神经退行性疾病的分子靶标和作用机理,并展望了这类化合物“老药新用”对抗神经退行性疾病的可能性。
1 3-取代吲哚酮类化合物具有神经保护活性,可能对抗神经退行性疾病
神经退行性疾病包括帕金森病(Parkinson’s disease,PD)、AD、亨廷顿病和肌萎缩侧索硬化症等。这些疾病的主要特征是大脑和脊髓神经元病理性丧失。由于神经元难以再生,因此神经损伤很难被逆转,损伤后可能导致神经系统渐进性功能障碍。神经退行性疾病的病因尚不清楚。一般认为多个靶点联合参与疾病发生发展:如遗传、环境等多种因素通过氧化应激、线粒体损伤、异常蛋白聚集等多种机制导致PD患者中脑多巴胺能神经元退行性死亡,随着多巴胺能神经元减少,患者出现静止性震颤、肌肉僵直、行动迟缓等运动障碍[4]。AD病因假说包括Aβ毒性假说、tau蛋白过度磷酸化假说、胆碱能下降假说等[5]。随着病程进展,AD患者大脑胆碱能神经元退行性丢失,出现渐进性记忆障碍、认知功能障碍、人格改变及语言障碍等症状[6]。

Tab 1 The neuroprotective mechanisms of 3-substitute indolines in treating neurodegenerative disorders
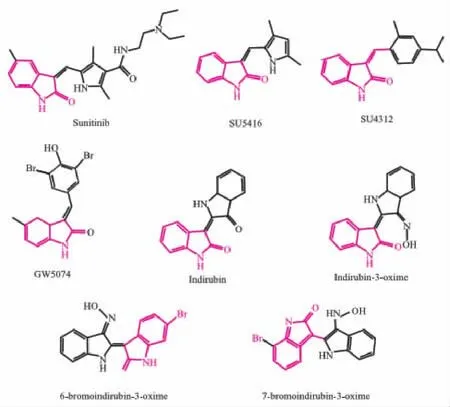
Fig 1 Chemical structures of representative 3-substitute indulines
近年来,包括我们的多个课题组分别独立发现,3-取代吲哚酮类化合物在体外培养神经元有显著神经保护作用。舒尼替尼、SU4312、SU5416在体外培养的SH-SY5Y细胞和小脑颗粒神经元能阻止1-甲基-4苯基吡啶离子(1-methyl-4-phenylpyridinium ion,MPP+)诱导的神经元凋亡[7];舒尼替尼、GW5074可对抗撤钾引起的小脑颗粒神经元凋亡[8];靛玉红-3-肟在PC12细胞能抑制6-羟基多巴胺(6-hydroxydopamine,6-OHDA)诱导的神经元死亡[9]。靛玉红-3-肟、7-溴-靛玉红-3-肟和舒尼替尼还能在SH-SY5Y细胞、小胶质细胞阻止Aβ诱导的细胞损伤。此外,药代动力学数据显示3-取代吲哚酮类化合物可以穿过血脑屏障,提示这些化合物可能有中枢神经系统药理活性。
3-取代吲哚酮类化合物在多种神经退行性疾病动物模型中也显示了极强的神经保护活性,阻止运动障碍和认知损伤。SU4312、SU5416在斑马鱼中可抑制1-甲基-4-苯基-1,2,3,6-四氢吡啶(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine,MPTP)诱导的运动障碍和中脑多巴胺能神经元死亡,提示其可能用于PD治疗[7, 8];舒尼替尼可对抗东莨菪碱诱导的小鼠认知障碍[10];靛玉红-3-肟和7-溴-靛玉红-3-肟能对抗侧脑室注射Aβ诱导的AD小鼠认知能力下降[11];此外,靛玉红和SU4312还能降低MPTP诱导的小鼠纹状体单胺类神经递质下降[12],对抗海人藻酸诱导的纹状体损伤[13]。这些研究均显示3-取代吲哚酮类化合物具有在体神经保护作用,可能用于治疗AD、PD等神经退行性疾病。
2 3-取代吲哚酮类化合物通过多靶点作用产生神经保护
3-取代吲哚酮类化合物产生神经保护作用的靶点是什么?是否与其RTK抑制活性有关?我们发现不具备3-取代吲哚-2酮结构的RTK抑制剂并不能产生类似的神经保护作用,提示3-取代吲哚酮类化合物神经保护作用可能不依赖其RTK抑制活性,而是通过作用其他靶点实现[7]。
c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)通路是神经元凋亡的重要调节通路,与神经元退行性丢失密切相关。活化的JNK蛋白可磷酸化下游转录因子c-Jun的Ser63和Ser73位点,激活c-Jun,进而诱导神经元凋亡。撤钾或双氧水刺激可通过激活JNK通路、磷酸化c-Jun,诱导小脑颗粒神经元凋亡;MPTP也可通过激活JNK通路诱导中脑多巴胺能神经元死亡。JNK抑制剂可阻止这些刺激诱导的神经元凋亡,显示抑制JNK通路具有神经保护作用。体外激酶活性实验显示舒尼替尼、靛玉红-3-肟等3-取代吲哚酮类化合物对JNK磷酸化有抑制作用。其中,靛玉红-3-肟对JNK三个亚型(JNK1、JNK2和JNK3)的半抑制剂量(IC50)分别为0.8、1.4和1.0 μmol·L-1[14]。靛玉红-3-肟在体外培养的小脑颗粒神经元和皮层神经元可剂量依赖性阻断撤钾以及秋水仙碱诱导的JNK通路激活、阻止神经元凋亡[8]。以上结果显示抑制JNK通路可能是3-取代吲哚酮类化合物实现神经保护的重要方式。
糖原合成酶激酶3β (glycogen synthase kinase-3β,GSK3β)也参与神经元退行性死亡。GSK3β是Aβ、MPTP等神经毒素产生毒性的关键激酶。这些神经毒素通过改变GSK3β磷酸化状态(抑制Ser9位点磷酸化、促进Ser396位点磷酸化),增加GSK3β活性,诱导神经元凋亡。此外,GSK3β还可抑制神经干细胞增殖。因此,GSK3β抑制剂可通过促进神经元存活和神经干细胞分化联合产生神经保护。舒尼替尼、GW5074、靛玉红、靛玉红-3-肟等3-取代吲哚酮类化合物可直接抑制GSK3β。其中,靛玉红对GSK3β的抑制有高度选择性,可有效逆转GSK3β活性增加诱导的tau蛋白磷酸化和细胞凋亡[15-16]。靛玉红-3-肟抑制GSK3β的IC50更达22 nmol·L-1[17]。靛玉红-3-肟通过降低GSK3β活性,阻止MPTP诱导的大鼠中脑多巴胺能神经元死亡,改善动物运动能力[11, 16]。6-溴-靛玉红-3-肟可通过抑制GSK3β剂量依赖性地降低tau蛋白水平和tau蛋白的磷酸化,其IC50为1.5 μmol·L-1[18]。此外,靛玉红-3-肟和7-溴-靛玉红-3-肟可通过抑制GSK3β,降低Aβ诱导的tau蛋白异常磷酸化,阻止高脂饮食诱导的小鼠认知障碍[17, 19-20]。因此,GSK3β也是3-取代吲哚酮类化合物神经保护作用的重要靶标。
细胞周期素依赖性激酶(cyclin-dependent kinases,CDK)是调节细胞周期的关键蛋白。其中CDK5在神经退行性疾病中有重要作用。CDK5活性在AD、PD的啮齿类模型显著增加,活化的CDK5可通过促进Aβ、tau、α-synuclein等疾病相关蛋白异常聚集,诱导神经毒性,促进神经元死亡。舒尼替尼、靛玉红、靛玉红-3-肟等3-取代吲哚酮类化合物均可直接抑制CDK5活性,其中,舒尼替尼抑制CDK5的IC50为4.2 μmol·L-1[21]。舒尼替尼在小鼠海马神经前体细胞中可抑制HIV外膜糖蛋白gp120引起的CDK5活化,阻止神经元死亡[21]。舒尼替尼在gp120转基因小鼠还能阻止tau蛋白异常磷酸化,预防神经元退行性死亡,提高动物认知能力[21]。靛玉红-3-肟不仅是有效的CDK抑制剂,而且毒性低、分子量小、有较好的细胞膜通透性,可以改善AD大鼠的学习记忆功能,减少Aβ诱导的额颞叶皮层和海马CA1区神经元凋亡[11]。此外,舒尼替尼、靛玉红-3-肟通过抑制CDK5,可阻止tau和DARPP-32等蛋白异常磷酸化,产生神经保护[11]。由此可见,CDK5也是3-取代吲哚酮类化合物实现神经保护的重要靶点。
肌细胞增强因子2(myocyte enhancer factor-2,MEF2)是促进神经元存活的重要转录因子。MEF2转录活性的调节机制复杂,在神经元细胞质中,MEF2可被GSK3β、CDK5等激酶修饰。经修饰的MEF2难以入核,无法执行转录功能。Aβ、MPTP、6OHDA等神经毒素可通过促进MEF2失活诱导神经元死亡。靛玉红-3-肟等3-取代吲哚酮类化合物除直接抑制GSK3β、CDK5、JNK等激酶活性外,还能直接增强MEF2转录活性,促进SH-SY5Y细胞存活[22]。SU4312可通过直接增强MEF2转录活性逆转MPTP诱导的黑质多巴胺能神经元损伤,抑制氧化反应,维持线粒体功能,阻止多巴胺及其代谢物的消耗,抑制大鼠运动能力下降[13]。
一氧化氮合酶(nitric oxidase synthase,NOS)在神经元退行性死亡中也发挥关键作用。神经元性一氧化氮合酶(neuronal NOS,nNOS)过度激活可产生过量NO,进而诱导神经元死亡。Aβ诱导的内源性NO水平升高是AD早期神经元死亡的主要原因。NO还参与了MPP+/MPTP等PD相关神经毒素诱导的神经元损伤。舒尼替尼、SU4312和SU5416在体外可直接抑制nNOS活性,IC50分别为9.6、22.7、19.0 μmol·L-1。分子对接实验显示3-取代吲哚酮类化合物可能通过其吲哚-2-酮母核与nNOS的heme基团相互作用,抑制nNOS。这些化合物在小脑颗粒神经元可通过抑制nNOS活性,阻止撤钾及MMP+等刺激诱导的NO水平上升和神经元死亡[7, 23]。此外,靛玉红-3-肟还可通过降低NO水平,阻止细菌脂多糖诱导的小胶质细胞损伤[24]。以上结果显示3-取代吲哚酮类化合物可通过抑制nNOS、降低NO水平产生神经保护。
我们最近研究还发现,3-取代吲哚酮类化合物可通过抑制神经递质代谢过程中重要蛋白发挥抗神经退行性疾病的作用,如舒尼替尼可竞争性抑制乙酰胆碱酯酶(acetylcholinesterase,AChE),其IC50低于1 μmol·L-1,可有效阻止脑内乙酰胆碱分解,对抗东莨菪碱诱导的小鼠认知障碍[12]。SU4312能选择性抑制单胺氧化酶-B(monoamine oxidase-B,MAO-B),其IC50值为0.2 μmol·L-1[13]。显示这类化合物还可能通过调节神经递质系统产生神经保护作用。
3 3-取代吲哚酮类化合物可望发展为新型抗神经退行性疾病药物
综上所述,3-取代吲哚酮类化合物可通过JNK、GSK-3β、CDK5、MEF2、nNOS、AChE、MAO-B等多靶点实现神经保护(Fig 2)。由于神经退行性疾病与多因素相关,传统的单靶点药物很难阻止神经元死亡,实现对因治疗。3-取代吲哚酮类化合物这样的多靶点神经保护药物可能是治疗神经退行性疾病的重要方向。另一方面临床药物安全性确定,且被临床医生熟知。因此在“老药新用”的研发可免除毒理学和药代动力学评价,较新药研发大大缩短了成本和时间。3-取代吲哚酮类化合物作为临床抗肿瘤药物“老药新用”开发为治疗神经退行性疾病具有极大的优势,对其神经保护作用及其机制的进一步研究有望获得一类新型抗神经退行性疾病药物。
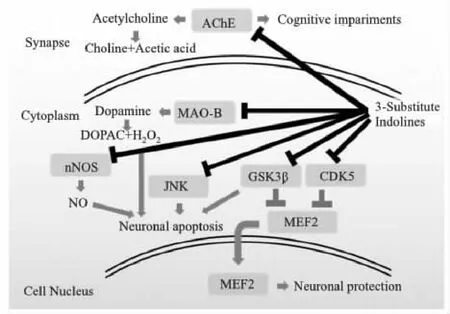
Fig 2 Main anti-neurodegeneration targets of 3-substituted indolones
