探索DNA条形码对哈摇蚊类群部分属物种鉴定的有效性
2020-10-14余海军黄鑫张钰莹周晶晶田召志陈佳林王茜
余海军,黄鑫,张钰莹,周晶晶,田召志,陈佳林,王茜
探索DNA条形码对哈摇蚊类群部分属物种鉴定的有效性
余海军,黄鑫,张钰莹,周晶晶,田召志,陈佳林,王茜通信作者
(天津农学院 水产学院 天津市水生生态及养殖重点实验室,天津 300384)
采集中国部分地区哈摇蚊类群,并对其进行了DNA条形码的鉴别研究。研究结果显示:种间平均遗传距离大约是种内平均遗传距离的2.8倍,种内与种间存在明显的“DNA barcoding gap”。基于Automatic Barcode Gap Discovery(ABGD)法分析遗传距离与其分布频数的关系;邻接树(Neighbor joining tree,NJ tree)分析结果指出,87.5%的物种能区分开,并与形态学鉴定一致。本研究为探索DNA条形码在哈摇蚊类群中鉴别的有效性提供了基础数据,同时为填补中国摇蚊昆虫的DNA条形码数据提供丰富的材料。
哈摇蚊类群;DNA条形码;物种鉴定;遗传距离
摇蚊因大部分幼虫体色为红色而常被渔民们称为“红虫”,其隶属于双翅目(Diptera)长角亚目(Nematocera),其同时也是水生态系统中丰富度极高的无脊椎动物类群[1],能生存于各种类型的水体[2]。由于不同的摇蚊类群对不同水域环境有一定的适应性和敏感性,因此可以不同摇蚊物种的分布为指标,监测水体水质变化[3]。
哈摇蚊群归属于摇蚊亚科,其类群包括哈摇蚊属()、枝角摇蚊属()、拟枝角摇蚊属()、小摇蚊属()、隐摇蚊属()、拟隐摇蚊属()、弯铗摇蚊属()和罗摇蚊属()8个近缘属[4]。该类群的形态鉴别特征以生殖节上的下附器退化为特征,其中、是上下附器都比较退化,其余6属的上附器为细长状、足靴状、指状和叶状等形状而未退化。尽管属间的分类特征明显,但是各属之间的物种存在极大的相似性,从而导致形态学鉴定难度加大。
2003年加拿大动物学家Hebert提出了DNA条形码,即通过使用一段标准的简短基因序列作为条形码对物种进行鉴别[5]。近年来DNA条形码已成功用于摇蚊昆虫的物种鉴定,特别是在物种的区分上,Lin等人基于DNA条形码对长跗摇蚊属()121个形态种进行物种分析,结果表明,DNA条形码有效区分了早期形态学研究分类中94.6%的长跗摇蚊属种类[6]。Song等通过DNA条形码对多足摇蚊属()的93个形态种进行物种鉴别,结果显示94.4%的多足摇蚊属物种能通过DNA条形码区分[7]。此外,在新种的发现上,DNA条形码也起到重要的验证作用,Song等人基于DNA条形码对一个中国新纪录属进行了物种鉴别,结果显示该属单独聚为一支,与形态学研究结果相符[8]。Qi等人通过DNA条形码对一个在鱼塘表面滑行的海洋物种进行了物种鉴别,结果显示其与二叉摇蚊属()聚为一支,验证了其属于摇蚊的形态学鉴定,并将该物种命名为Qi& Lin sp. n,证明了摇蚊不只是生存在淡水环境中,也可以在海洋环境中生存[9]。Lin等人通过DNA条形码对sp. n.进行分子分析,结果显示其能与其他多足摇蚊属物种区分开,与形态学鉴定一致[10]。
本研究通过对中国部分地区的哈摇蚊类群物种中8属20种(总计40样本),选用COI基因作为分子标记,进行了DNA条形码研究,研究结果为国内摇蚊昆虫的DNA条形码数据库提供基础数据,并对国内外的同类研究做一个补充。
1 材料与方法
1.1 样品来源
本研究所选对象为哈摇蚊类群物种中的8属20种,均来自中国部分地区野外采集,采集方式为扫网和灯诱,标本均保存于75%~85%乙醇中。所有采集的样本均在解剖镜下解剖封片,并将头部和胸部保存于85%乙醇中,用于后续分子研究,所有证据标本均保存于天津农学院水产遗传育种实验室。研究中的对比序列为30条,均从BOLD(Barcode Of Life Data)数据库上下载。
1.2 DNA提取、PCR扩增、序列对比和分析
1.2.1 DNA提取
本试验采用的是试剂盒(QIAGEN DNeasy Blood and Tissue kit)提取法,将整个胸部和头部经蛋白酶处理后,按照试剂盒说明书提取样本DNA。
1.2.2 PCR扩增
聚合酶链式反应(PCR)扩增使用COI基因通用引物LCO1490和HCO2198[11]。反应体系为25 μL,包含12.5 μL 2× EsMasterMix(CoWin Biotech Co,Beijing,China),上下游引物各0.625 μL,dd H2O 8.75 μL。PCR通过MasterCycler Gradient进行扩增,扩增程序如下,预变性98 ℃ 10s,94 ℃ 5 min,35个循环:94 ℃ 1 min,51 ℃ 1 min,72 ℃ 1 min。最后延伸72 ℃ 5 min,10 ℃保存。PCR扩增产物经1.5%琼脂糖凝胶电泳检测后送北京六合华大基因科技股份有限公司进行测序。
1.3 数据对比和分析方法
1.3.1 序列对比与序列特征分析
将测序公司返回的测序序列导入SeqMan中进行序列编辑,将编辑好的序列导出为FASTA文件保存,然后导入MEGA7.0进行比对并转换成氨基酸检查序列检查是否存在终止密码子[12],并对序列特征及各碱基含量进行统计。
1.3.2 基于遗传距离分析
基于DNA条形码遗传距离的分析方法有ABGD法。利用ABGD[13]软件在线分析,将编辑好的FASTA文件在线提交到ABGD网站(http:// wwwabi.snv.jussieu.fr/public/abgd/),选择Kimura- 2-parameter(K2P)模型,种内差异先验值(Prior intraspecific divergence)为0.001到0.100,相对gap宽度值(relative gap width)为1.0,其余参数设置为默认值[6]。
1.3.3 基于系统发育树分析
在建树过程中,将比对好的FASTA文件导入MEGA 7.0中并选用K2P分子进化模型,节点处置信度为1 000,其他设置均为默认值。
单倍型结构分析通过将比对好的NEXUS文件导入PopART软件选用TCS模型分析[14-15]。
2 结果与分析
2.1 序列特征结果
本试验一共获得40条COI序列。所有序列的长度均为658 bp,各序列特征所示,其中各碱基含量的平均值分别为A=27.5%,T=38.0%,C=18.1%,G=16.5%,序列存在碱基的偏倚性,C+G的含量显著低于A+T的含量,特别是第三核酸位点A+T的含量高达88.4%。通过比对序列得出,C(保守位点)占63.5%,V(变异位点)占36.2%。大部分的变异位点都集中在第三核酸位点,其比例为84.4%。结果表明,在第三核酸位点上的碱基进化速率快。
2.2 遗传距离结果
本试验40个样本,形态鉴定为种,种内遗传距离为0~0.154 4,平均值为0.048 4;种间遗传距离为0.068 8~0.173 3,平均值为0.137 8。
ABGD能使用直方图来展现种内、种间遗传距离及其分布频数的关系,比较种内和种间的遗传距离,当种间最小遗传距离大于种内最大遗传距离时,则出现“空白区”,如图1所示。
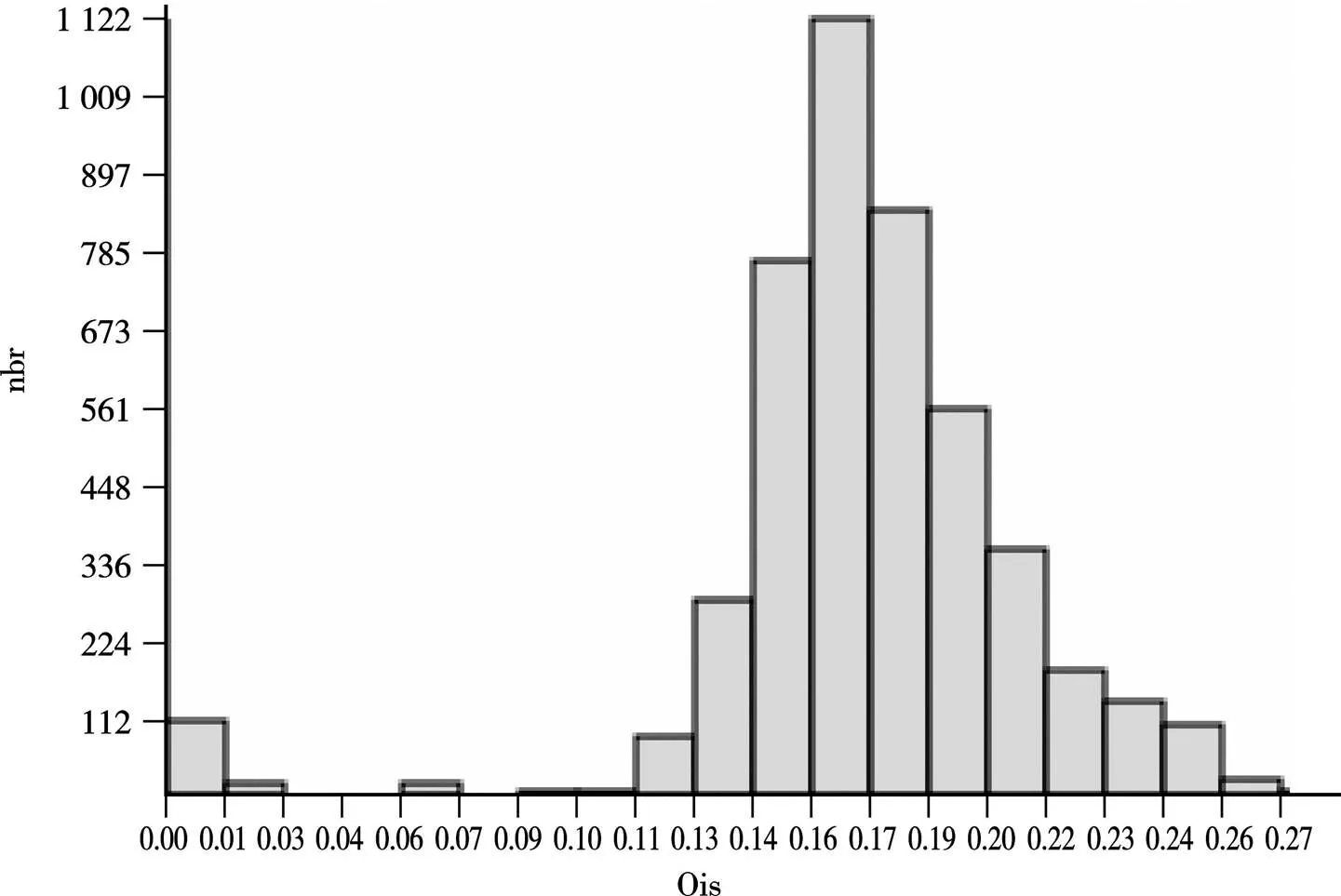
图1 摇蚊亚科物种遗传距离频率直方图
2.3 系统发育树结果
基于40条DNA条形码序列,构建NJ树,如图2所示。

图2 哈摇蚊类群NJ树
注:不同颜色或形状代表不同物种,下同
3 讨论
3.1 基于遗传距离阈值分析
DNA条形码研究中,种内与种间遗传距离的差异是一项非常重要的指标,当种间遗传距离大于种内遗传距离时,就会形成一个明显的空白区,即“DNA barcoding gap”。本研究中种内平均遗传距离为0.048 4,种间平均遗传距离为0.137 8,后者大约是前者的2.8倍,形成了“DNA barcoding gap”。
遗传距离阈值是指物种在能形成“DNA barcoding gap”的前提下,使用物种种间遗传距离与参比的遗传距离阈值进行对比,当种间遗传距离大于参比的阈值时,则可以将物种区分为不同物种。阈值法在DNA条形码的物种鉴定中起着不可替代的作用,然而随着DNA条形码研究不断深入,有学者发现不同物种DNA条形码的阈值各不相同,没有一个统一的标准阈值[16]。BOLD数据库最初将遗传距离1%作为生物物种的划分阈值,由于其阈值宽泛,在一定程度上造成了物种的“鉴别过度”,导致同种异名的出现,为了不使物种混乱,近年来BOLD系统默认鉴别阈值为3%[17]。
将本研究的序列结果导入Speciesdentifier软件中,设定阈值为1%~10%,物种条形码聚类结果如图3所示,形态鉴定的数量为20种,当阈值在7%~8%时,mOTU数量最接近形态鉴定的数量,我们推测哈摇蚊类群的遗传距离阈值为8%~9%。关于摇蚊亚科中其他类群的阈值,Song等区分多足摇蚊属的遗传距离阈值为7%~9%[7],Lin等人区分长跗摇蚊属的遗传距离阈值为4%~5%[6]。综上所述,本研究分析的哈摇蚊类群遗传距离阈值,与其同亚科的类群遗传距离阈值相比,没有较大差异,在一定程度上证明了DNA条形码能有效地对哈摇蚊群部分属物种进行鉴定。

图3 基于阈值的物种聚类分析
3.2 基于系统发育树分析
系统发育树能直观地反映物种之间的亲缘关系,本研究中采用的是NJ法,其基于遗传距离和最小进化原理,通过自举检验法进行聚类,并通过统计给定树分支的可信度,得出最优树[18]。在NJ树中,87.5%的物种能区分开,与形态学鉴定结果一致。本研究显示在哈摇蚊属中(图4),双叶哈摇蚊()与参考序列分成了3支,但因不能查询参考序列的证据标本,所以无法对该数据做出明确的定论;此外,在哈摇蚊属中,TN01短叶哈摇蚊()与参考序列GBMIN55160-17长距哈摇蚊()聚为一支,且置信度为100%,据闫春财博士论文中描述,与在外观形态上很相似,即使通过生殖节的比对也不易区分,但腋瓣上是否具有缘毛可区分二者[19],TN01经过形态鉴定此物种应为(图5),而非。基于以上推断,对研究中哈摇蚊属所有个体的DNA条形码数据在PopART软件中使用TCS模型进行单倍型结构分析,结果如图6所示:除与聚为一体外,其他个体都能单独聚为一体,该结果表明TN01与GBMIN55160-17出现同物异名。由于数据库中GBMIN55160-17参考序列不能提供证据标本与TN01比较,本研究无法做出准确的定论,在今后的研究中将进一步验证。系统发育树的结果在一定程度上表明,DNA条形码能有效鉴别摇蚊亚科物种。研究者在进行摇蚊亚科昆虫遗传距离界定时,需要比对正确而无疑的DNA条形码数据,在今后的工作中,项目组将与条形码的同行做更深入地交流,为中国摇蚊亚科DNA条形码数据库的完善提供参考。
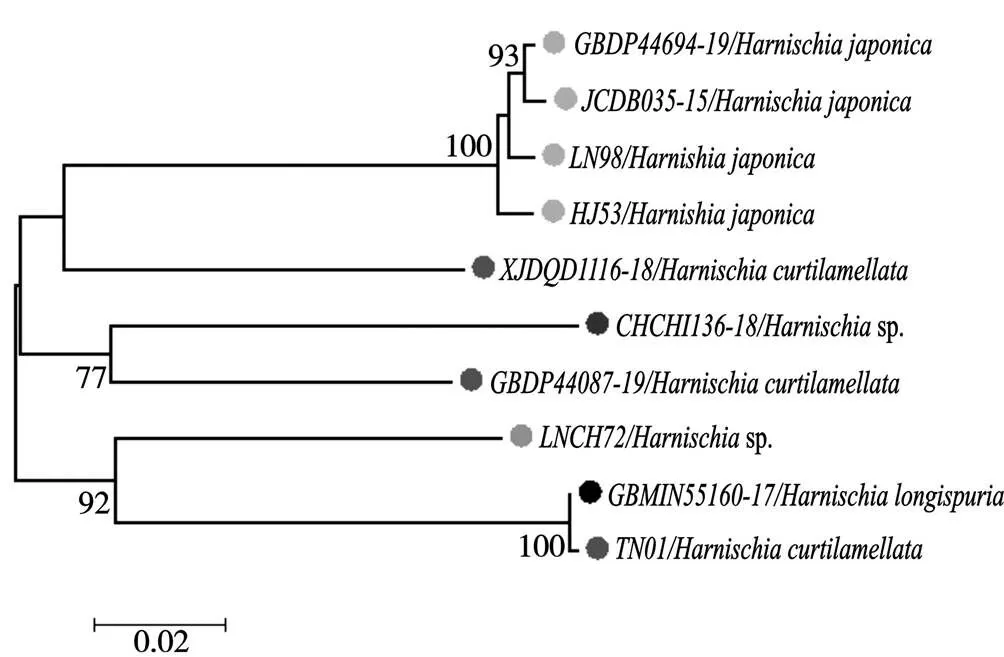
图4 哈摇蚊属物种NJ树

图5 TN01样本的证据标本(翅部分)
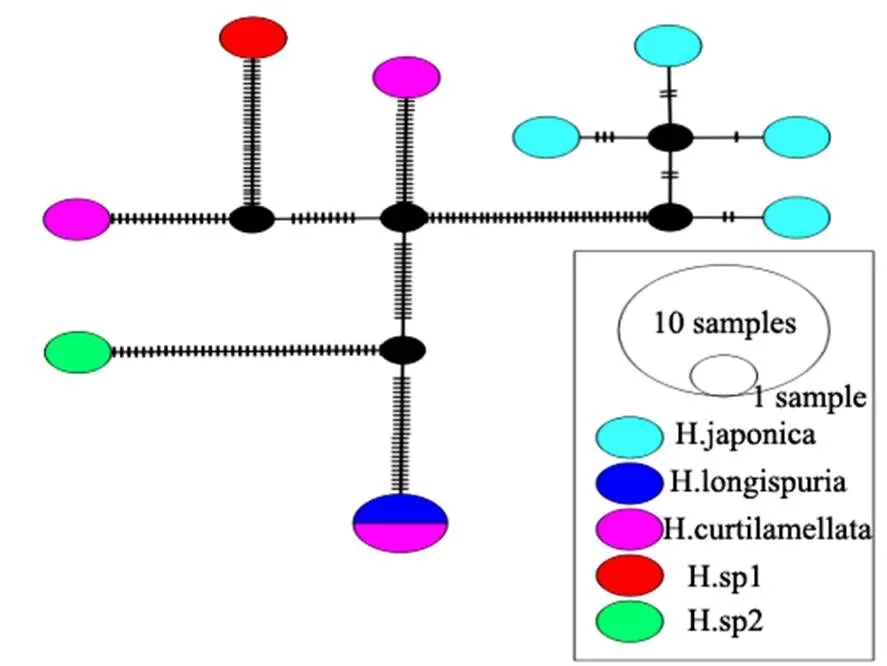
图6 哈摇蚊属基于TCS haplotype network的分析
4 结论
本研究通过采集中国部分地区的哈摇蚊类群昆虫8属20种(共计40样本),进行了DNA条形码数据分析,运用ABDG法分析该数据集种内、种间遗传距离及其分布频数的关系,基于NJ树结果得出数据集分离出20个物种,与传统形态学鉴定一致,证明DNA条形码能准确鉴别哈摇蚊类群昆虫,同时为填补中国摇蚊昆虫的DNA条形码数据提供丰富的材料。
[1] MiloševićD,SimićV,StojkovićM,et al. Chironomid faunal composition represented by taxonomic distinctness index reveals environmental change in a lotic system over three decades[J]. Hydrobiologia,2012,683(1):69-82.
[2] Nicacio G,Juen L. Chironomids as indicators in freshwater ecosystems:An assessment of the literature[J]. Insect Conservation and Diversity,2015,8(5):393-403.
[3] Sæther O A. Zoogeographical patterns in Chironomidae(Diptera)[J]. SIL Proceedings,2000,27(1):290-302.
[4] 王新华,郑乐怡,纪炳纯. 中国摇蚊亚科记述Ⅲ. 哈摇蚊属(双翅目:摇蚊科)[J]. 动物分类学报,1993,18(4):459-465.
[5] Hebert P D N,Cywinska A,Ball S L,et al. Biological identifications through DNA barcodes[J]. Proc R Soc Lond B Biol Sci,2003,270(1512):313-321.
[6] Lin X L,Stur E,Ekrem T. Exploring genetic divergence in a species-rich insect genus using 2790 DNA barcodes[J]. Plos One,2015,10(9):1-24.
[7] Song C,Lin X L,Wang Q,et al. DNA barcodes successfully delimit morphospecies in a superdiverse insect genus[J]. Zoological Scripta,2018,47(3):311-324.
[8] Song C,Lin W B,Zhang R L,et al. Two gynandromorphic species ofKieffer,1912(Diptera:Chironomidae),with DNA barcodes from Oriental China[J]. The Pan-pacific Entomologist,2017,93(2):1-10.
[9] Qi X,Lin X L,Ekrem T,et al. A new surface gliding species of Chironomidae:An independent invasion of marine environments and its evolutionary implications[J]. Zoological Scripta,2019,48(1):81-92.
[10] Lin X L,Yu H J,Zhang R L,et al.()sp.n.(Diptera:Chironomidae)from Gaoligong Mountains,Yunnan,China[J]. Zootaxa,2019,4571(2):255-262.
[11] Folmer O,Black M,Hoeh W,et al. DNA primers for amplification of mitochondrial cytochrome coxidase subunit 1 from diverse metazoan invertebrates[J]. Molecular Marine Biology and Biotechnology,1994,3(5):294-299.
[12] Kumar S,Stecher G,Tamura K. MEGA7:Molecular evolutionary genetics analysis version 7.0 for bigger datasets[J]. Molecular Biology and Evolution,2016,33(7):1870-1874.
[13] Puillandre N,Lambert A,Brouillet S,et al. ABGD,automatic barcode gap discovery for primary species delimitation[J]. Molecular Ecology,2012,21(8):1864-1877.
[14] Clement M,Posada D,Crandall K A. TCS:A computer program to estimate gene genealogies[J]. Molecular Ecology,2000,9(10):1657-1659.
[15] Leigh J W,Bryant D. POPART:Full-feature software for haplotype network construction[J]. Methods in Ecology and Evolution,2015,6(9):1110-1116.
[16] Collins R A,Cruickshank R H. The seven deadly sins of DNA barcoding[J]. Molecular Ecology Resources,2013,13(6):969-975.
[17] Ratnasingham S,Hebert P D N. BOLD:The barcode of life data system[J]. Molecular Ecology Notes,2007,7(3):355-364.
[18] Duarte S,Batista D,Barlocher F,et al. Some new DNA barcodes of aquatic hyphomycete species[J]. Mycoscience,2015,56(1):102-108.
[19] 闫春财. 中印区哈摇蚊属复合体系统学研究(双翅目:摇蚊科)[D]. 天津:南开大学,2005.
Exploring the utility of DNA barcoding in species identification ofcomplex
YU Hai-jun, HUANG Xin, ZHANG Yu-ying, ZHOU Jing-jing, TIAN Zhao-zhi, CHEN Jia-lin, WANG QianCorresponding Author
(Key Laboratory of Aquatic-Ecology and Aquaculture of Tianjin, College of Fisheries, Tianjin Agricultural University, Tianjin 300384, China)
This study collected Harischia complex species in regions of China, using DNA barcoding for species identification. The results showed: the mean interspecific genetic distance was 2.8 times larger than the mean intraspecific genetic distance, with existing distinct “DNA barcoding gap”. Based on Automatic Barcode Gap Discovery genetic distance was analyzed, and it was related with distributed frequency. The results of Neighbor joining tree analysis pointed that 87.5%of the species were distinguished, and it kept conformed to morphological identification. Our results produced basal data for exploring the utility of DNA barcoding in species identification ofcomplex, and provided abundant material for filling up the Chinese DNA barcodes database of Chironomidae.
complex; DNA barcoding; species identification; genetic distance
1008-5394(2020)03-0057-05
10.19640/j.cnki.jtau.2020.03.013
S931.1
A
2019-06-26
国家自然科学基金(31672264,31672324)
余海军(1994-),男,硕士在读,主要从事分子系统学研究。E-mail:yhj18322040463@163.com。
王茜(1971-),女,副教授,博士,主要从事水生动物及昆虫分子系统学的研究。E-mail:wqgt1999@163.com。
责任编辑:张爱婷
