基于R软件和数据库的生物信息学分析设计
2020-07-31张婕李梦婷
张婕 李梦婷

摘 要:选取NCBI基因表达谱数据库中访问号为GSE41439的基因芯片数据集为分析对象,首先利用R软件筛选差异表达基因并绘制成聚类热图,然后将差异基因上传至DAVID数据库进行GO功能与KEGG通路富集分析,接着利用STRING数据库构建蛋白质互作网络,并利用Cytoscape软件进行可视化,以直观地观察蛋白与蛋白之间的相互关系。由蛋白互作网络筛选出4个关键基因:PIK3R1、GNAS、GNAL、GNG4,可对其进行更深入的讨论。此方法适用于多种基因芯片的研究,具有很好的可推广性,将其运用于疾病相关的基因芯片,可为医学诊断与精准治疗提供一定的帮助。
关键词:生物信息学;R软件;DAVID数据库;STRING数据库;Cytoscape
中图分类号:R319 文献标识码:A 文章编号:2096-4706(2020)04-0076-04
Abstract:The gene chip data set with access number GSE41439 in NCBI gene expression profile database is selected as the analysis object. Firstly,the differential expression genes are screened by R-studio and the clustering heat map is drawn,then the differential genes are uploaded to DAVID database for GO function and KEGG pathway enrichment analysis,and then the protein interaction network is constructed by using STRING database,and can be seen by using Cytoscape software to observe the relationship between protein and protein directly. Four key genes,PIK3R1,GNAS,GNAL and GNG4,were screened out by protein interaction network,which can be further discussed. This method is suitable for the research of many kinds of gene chips,and has good generalization. It can be applied to the disease-related gene chips,which can provide some help for medical diagnosis and precise treatment.
Keywords:bioinformatics;R-studio;DAVID data base;STRING data base;Cytoscape
0 引 言
随着精准医疗与计算机技术的迅速发展,计算机技术在数据挖掘方面的优势逐渐显现,同时基因组学和蛋白质组学的快速发展积累了大量的生物数据,生物与计算机的结合让生命科学领域进入大数据时代[1]。生物信息数据库具有种类多、规模大、覆盖面广以及更新速度快等特点,充分利用这一特点,可以识别疾病的潜在治疗靶基因,挖掘基因的功能以及基因之间的关联性,为疾病的预防和治疗提供新的途径[2]。本文以NCBI高通量基因表达谱数据库(GEO)中访问号为GSE41439的基因芯片数据集为例,介绍基于R软件和数据库的生物信息分析方法,挖掘芯片所包含的潜在信息。该芯片基于GPL570平台,含有8个样本信息,比较了正常人胚胎干细胞系VUB01、VUB02、VUB03和VUB07及其含有20q11.21重复序列的亚系的基因表达差异。20q11.21的增加是染色体异常的一种,分析具有正常核型的人胚胎干细胞与获得20q11.21重复后的细胞内差异表达基因,可以为识别导致染色体异常的关键基因及其所参与的功能提供帮助。
1 基于R软件的基因芯片数据处理与初步分析
1.1 安装程序包
R软件是专业的统计软件,是统计计算、数据可视化的优秀工具,同时R也是免费开源的软件,在其官网和镜像网站中可以下载安装程序、源代码和程序包等[3]。R软件为用户提供了大量的程序包,使得用户能够灵活地运用这些程序包进行数据的分析及可视化,运用R软件处理基因芯片的第一步即是安装自己所需的程序包。
1.2 數据过滤及标准化
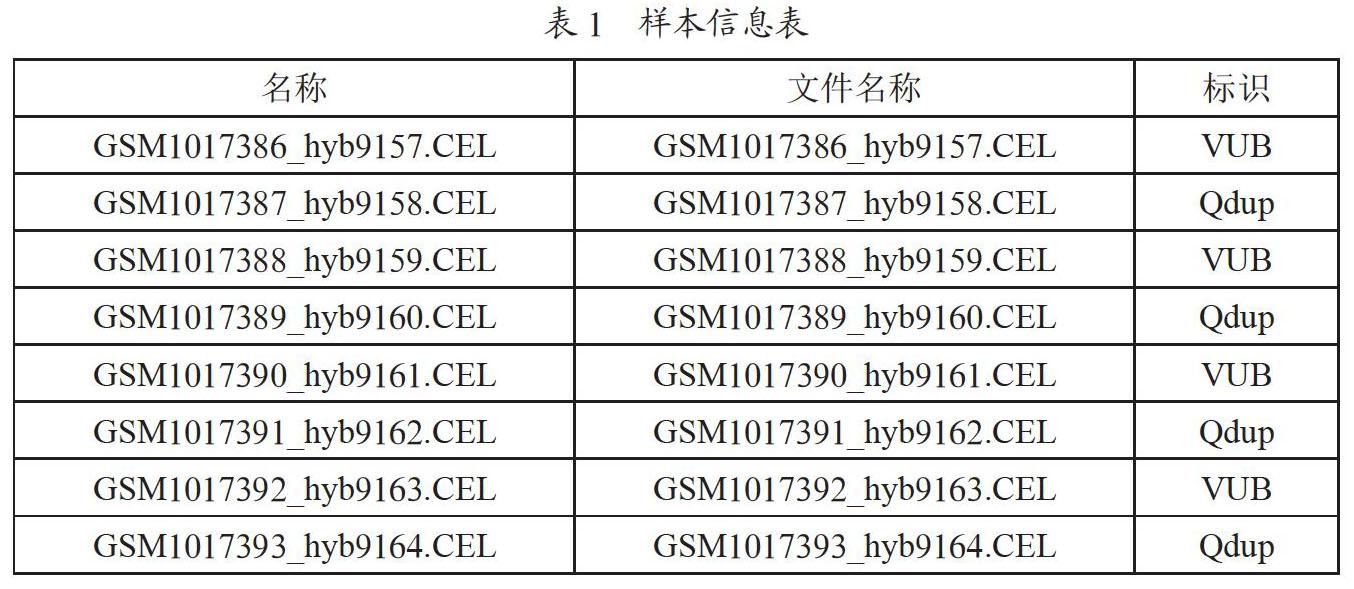
GEO数据库提供了大量开放共享的基因芯片数据集,分析芯片所包含的信息使得我们能够从分子层面认识样本,从而获取其中的关键基因,甚至可以作为疾病分子诊断与治疗的依据。从GEO数据库中下载访问号为GSE41439的基因芯片原始数据,并将其解压为CEL文件,整理其所包含的样本信息为如表1所示。
其中,名称为样本的名字,文件名称为样本文件的名字,标识为样本的标签与类型,各列之间以Tab键进行分隔,将整理好的样本信息文件,与解压好的CEL文件共同存于同一文件夹下,即可运用R软件的GC-RMA算法对其进行数据过滤及标准化。
1.3 筛选差异表达基因
差异表达基因是分析样本之间差异信息并进一步寻找核心基因的关键,R软件的limma包提供了相对完善的差异分析工具,本文即运用R软件的limma包进行差异表达基因的筛选,选定筛选条件为|logFC|>1.00且P.Value<0.05,进一步分析基因芯片蕴含的丰富信息,最终获得3个有意义的文件,分别为差异表达基因的分析结果、上调基因的具体结果以及下调基因的具体结果,文件自动存入默认工作路径下。
1.4 层次聚类热图绘制
层次聚类热图可以用于判断不同条件下的差异基因表达模式,直观地展示基因芯片的分析结果即某一个位置基因表达水平的高低,从而看出各差异基因在各样本中的表达情况。首先从GEO数据库下载GSE41439芯片的基因表达矩阵,并与通过R软件筛选到的差异表达基因进行整合,得到各差异基因在各个样本之间的表达矩阵。然后利用R软件对差异基因表达矩阵进行可视化,采用双向聚类的方法,根据某一样本中不同基因的表达水平将基因进行聚类,同时根据某一基因在不同样本中的表达水平将样本进行聚类,对基因在行方向进行标准化,设置行列方向的树高分别为100和20,同时选用由深到浅的颜色进行标记,绘制成层次聚类热图,如图1所示。
2 基于数据库的基因芯片数据挖掘
2.1 DAVID数据库进行富集分析
DAVID[4]是一个为大量基因列表提供一整套功能性注释的数据库,其从上传的基因列表中系统地提取具有生物意义的基因或蛋白,列出涉及到的疾病、蛋白功能域、GO功能、KEGG通路等。GO功能富集分析以及KEGG代谢通路富集分析可以帮助我们从分子层面更深入的了解差异表达基因以及它们之间的富集关系,从而找到富集差异基因的GO分类条目和KEGG通路,得出差异基因可能参与的基因功能以及代谢通路。
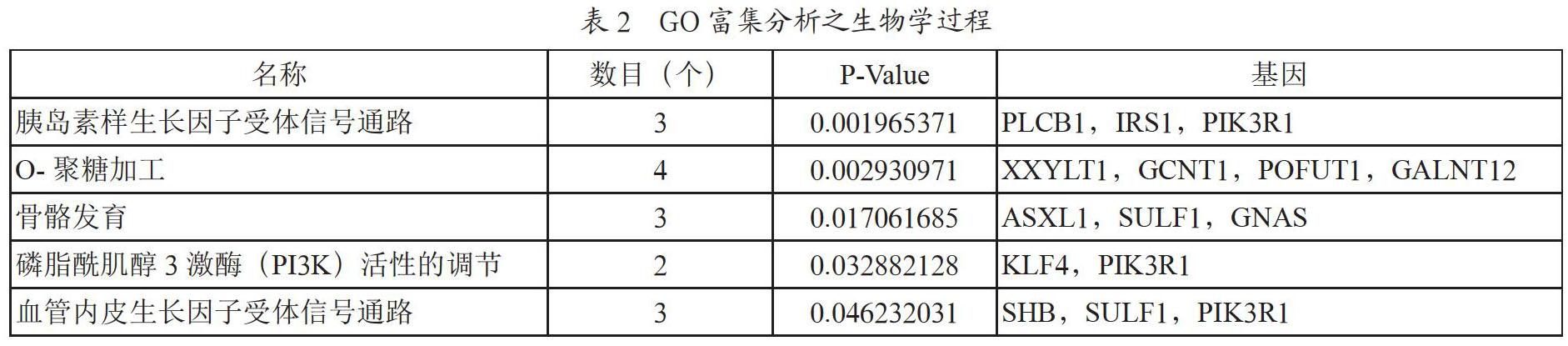
将差异表达基因名上传至DAVID在线数据库,并选择物种背景为homo sapiens,进行富集分析。设定p<0.05,将所得的差异基因归类到生物学过程(如表2所示)、分子功能、细胞组分以及KEGG通路三种生物学关系中,并将富集分析结果下载以便后续的可视化分析。
2.2 STRING数据库进行互作分析
STRING 11.0[5]数据库能够提供对蛋白质相互作用网络分析和预测的全局视图。为了得到差异表达基因之间的相互作用,我们将显著差异基因上传至STRING 11.0版在线数据库,并选择综合得分≥0.4的基因进行蛋白交互网络(PPI)构建。将没有相互作用的节点隐藏,最终得到共有48个节点和55条边的PPI网络,如图2所示,并导出其相互作用表格、蛋白序列以及注释等信息,以便后续的可视化分析。
3 数据库结果可视化
3.1 富集结果可视化之气泡图
气泡图可以直观的表征功能富集分析的结果,其中横轴代表基因比例,即条目所包含基因占所有基因的百分比,单位为%,纵轴代表GO富集分析的具体条目,点的大小反映基因的个数,而颜色的深浅反映P值的高低。本文将DAVID数据库分析所得的生物学过程富集结果导入R软件绘制成气泡图,如图3所示。
3.2 互作网络可视化之Cytoscape
Cytoscape是一个基于Java技术的开放源代码的网络可视化软件平台,主要用于复杂生物网络的分析研究设计,可以用其绘制基因表达调控网络、蛋白互作网络等任何与网络结构、层级有关系的内容[6]。Cytoscape软件可构建可视化的分子交互作用网络图,节点与节点的连线则表示彼此之间有相互作用,并可将已有的基因表达信息整合到网络图中,从而较为容易地观察蛋白与蛋白之间的关联性[7]。
本文将所得的相互作用表格、蛋白序列及注释信息等导入Cytoscape软件3.7.1版,构建可视化的交互网络。首先选择Cytoscape软件菜单“File-Import-Network from File”输入网络表格数据,并设置Source列和Target列及相关属性列,生成初步的调控网络。接着我们将其表达信息整合到网络的节点(Node)与边(Edge)中,通过选择Cytoscape软件控制面板“Control Panel”中的“Style”选项卡对节点、边和网络进行样式设置,其中每一个节点代表一个蛋白(基因),节点大小随度渐变,深色代表上调,浅色代表下调,每一条边代表一个交互关系,边的粗细随相互作用的强度渐变,最终获得可视化蛋白交互网络,如图4所示。
从图4中可以初步看出,整个交互网络以PIK3R1、GNAS、GNAL、GNG4为中心节点,与其他蛋白相互作用,其中PIK3R1、GNAS、GNAL显著上调,GNG4显著下调,这4个基因可能是导致20q11.21增加染色体异常的关键基因。GO功能富集分析结果表明这些关键基因与胰岛素样生长因子受体信号通路、骨骼发育、PI3K活性的调节、血管内皮生长因子受体信号通路等生物过程密切相关,且主要发挥胰岛素样生长因子受体结合、调节PI3K活性、信号传感器活动、调节跨膜受体蛋白酪氨酸激酶衔接活性等分子功能;KEGG通路富集分析结果表明差异基因显著富集到血清素能性突触传递通路、多巴胺能突触传递通路以及钙信号途径等,与染色体异常密切相关。我们可以初步猜测,20q11.21增加导致的染色体异常可能对这些富集到的生物过程、分子功能以及信号通路产生影响,有了初步的分析结果,则可以应用其他分析方法进一步探索并证明其中的分子机制,研究基因之间的关联性。
4 结 论
GEO数据库提供了大量与疾病相关的基因芯片信息,此研究方法能够使识别疾病潜在的治疗靶基因成为可能。在实际分析中,选取自己感兴趣的基因芯片数据集,运用R软件和生物信息相关的数据库对基因芯片的信息进行数据挖掘,并利用Cytoscape将其整合到网络图中,从而找出关键基因,分析其所参与的GO功能以及代谢通路。此外,也可将此数据存入数据库,以便在后续研究中调用和参考,为临床分子诊断和精准治疗提供一定的帮助。
参考文献:
[1] 褚皓.数据挖掘在生物信息学中的应用 [J].数字技术与应用,2018,36(10):123-124.
[2] LUSCOMBE NM,GREENBAUM D,GERSTEIN M. What is bioinformatics? A proposed definition and overview of the field [J]. Methods of Information in Medicine,2001,40(4):346-58.
[3] 吴剑,钱进.R软件在工科概率论与数理统计教学中的应用 [J].考试周刊,2019(29):29.
[4] HUANG D W,SHERMAN B T,QINA T,et al. DAVID Bioinformatics Resources:expanded annotation database and novel algorithms to better extract biology from large gene lists [J].Nucleic Acids Research,2007,35(Web Server issue):169-175.
[5] FRANCESCHINI A,SZKLARCZYK D,FRANKILD S,et al. STRING v9.1:protein-protein interaction networks,with increased coverage and integration [J].Nucleic Acids Research,2013,41(D1):808-815.
[6] 楊淼,杜菁,李冬果,等.基于Cytoscape的miRNA调控网络的构建与研究 [J].中国医学装备,2018,15(10):95-97.
[7] HAMMOND D E,HYDE R,KRATCHMAROVA I,et al. Quantitative Analysis of HGF and EGF-Dependent Phosphotyrosine Signaling Networks [J].Journal of Proteome Research,2010,9(5):2734-2742.
作者简介:张婕(1998.10-),女,汉族,江苏淮安人,本科在读,研究方向:生物信息学。
