非达霉素原料药中有关物质的检查方法研究
2020-07-09王娅莉刘月班璐李晓露程晓昆任风芝张雪霞
王娅莉 刘月 班璐 李晓露 程晓昆 任风芝 张雪霞
中圖分类号 R927.2 文献标志码 A 文章编号 1001-0408(2020)05-0581-05
DOI 10.6039/j.issn.1001-0408.2020.05.15
摘 要 目的:建立非达霉素原料药中有关物质的检查方法。方法:分别考察正相-紫外检测器色谱系统、反相-蒸发光散射检测器色谱系统、反相-紫外检测器色谱系统对非达霉素原料药中有关物质的检出能力,优选出最佳色谱系统。建立有关物质检查的高效液相色谱法,色谱柱为Agilent Eclipse XDB C18,流动相A为0.2%三乙胺缓冲液(pH 3.8)-乙腈(55 ∶ 45,V/V)、流动相B为0.2%三乙胺缓冲液(pH 3.8)-乙腈(20 ∶ 80,V/V)(梯度洗脱),流速为1.0 mL/min,检测波长为230 nm,柱温为35 ℃,进样量为10 ?L;以不加校正因子的主成分自身对照法计算有关物质含量。结果:非达霉素在正相-紫外检测器色谱系统下不能有效分离杂质C、F,在反相-蒸发光散射检测器色谱系统下仅能检出杂质C、D、E、F 4个杂质;在反相-紫外检测器色谱系统下可检出11个杂质,故以此系统进行有关物质检查。在方法学考察中,非达霉素检测质量浓度线性范围为0.5~20.0 μg/mL(R2=0.999 9);检测限为0.05 ng,定量限为0.15 ng;重复性、中间精密度试验的RSD均小于2.0%(n=6);平均回收率为98.4%(RSD为3.6%,n=9)。3批非达霉素原料药中杂质含量分别为0.53%、0.51%、0.51%。结论:非达霉素在反相-紫外检测器色谱系统下进行杂质检查效果最好;建立的有关物质检查方法专属性强、灵敏度高,可用于非达霉素原料药中有关物质的检查。
关键词 非达霉素;高效液相色谱法;正相色谱;反相色谱;紫外检测器;蒸发光散射检测器;主成分自身对照法;有关物质
Study on Detection Methods for Related Substances in Fidaxomicin Raw Material
WANG Yali,LIU Yue,BAN Lu,LI Xiaolu,CHENG Xiaokun,REN Fengzhi,ZHANG Xuexia(New Drug Research & Development Company of NCPC/National Engineering Research Center of Microbial Medicine/Hebei Industry Microbial Metabolic Engineering &Technology Research Center, Shijiazhuang 050015, China)
ABSTRACT OBJECTIVE: To establish the method for the determination of related substances in fidaxomicin raw material. METHODS: The detection ability of NP-HPLC-UV, RP-HPLC-ELSD and RP-HPLC-UV systems for the related substances in fidamycin raw material was investigated and the best chromatographic system was selected. The HPLC detection method for the related substances was established. The detection was performed on Agilent Eclipse XDB C18 column with mobile phase A consisted of 0.2% triethylamine buffer solution (pH 3.8)-acetonitrile (55 ∶ 45, V/V), mobile phase B consisted of 0.2% triethylamine buffer solution (pH 3.8)-acetonitrile (20 ∶ 80, V/V) at the flow rate of 1.0 mL/min (gradient elution); the detection wavelength was set at 230 nm, and column temperature was 35 ℃; the sample size was 10 ?L. Calculation of the content of related substances was principal component self-control method without correction factor. RESULTS: The impurities C and F could not be separated effectively in NP-HPLC-UV system. In RP-HPLC-ELSD system, only impurities C, D, E and F could be detected. In RP-HPLC-UV system, 11 impurities could be detected. In the study of methodology, the linear ranges were 0.5-20.0 μg/mL for fidaxomicin (R2=0.999 9); the LOD was 0.05 ng, LOQ was 0.15 ng; RSDs of reproducibility and intermediate precision tests were less than 2.0% (n=6); average recovery was 98.4% (RSD=3.6%, n=9). The sum of impurities in 3 batches of raw materials were 0.53%, 0.51%, 0.51%, respectively. CONCLUSIONS: The effect of detecting impurities by RP-HPLC-UV are the best. Established method is specific and sensitive, and can be used for the determination of related substance in fidaxomicin raw material.
KEYWORDS Fidaxomicin; HPLC; Normal phase chromatogram; Reverse phase chromatogram; UV detector; ELSD; Principal component self-control method; Related substance
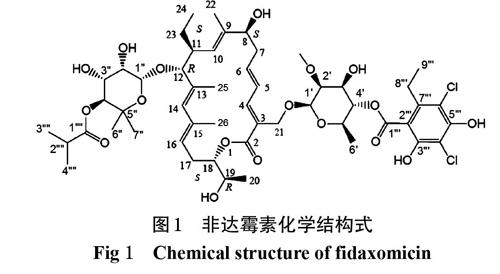
非达霉素(Fidaxomicin)又名非达米星,最早于1975年发现,是一种具有18元环复杂结构的大环内酯类抗生素,分子式为C52H74Cl2O18,分子量为1 058.04,对革兰氏阳性需氧及厌氧菌均具有较好的抗菌作用[1-4]。2011年被美国食品药品监督管理局(FDA)批准用于治疗艰难梭菌感染(CDI),且治疗CDI的效果优于现在临床常用的万古霉素[5-10]。
非达霉素为微生物的次级代谢产物,非达霉素粗品可由游动放线菌属菌株发酵产生,通过过滤、浸泡、浓缩、萃取、脱色、结晶、过滤、洗涤、干燥等一系列过程制得[11-12] 。非达霉素为台勾霉素B族的1种,其同系物多且结构极其相似[13-15]。在非达霉素发酵产生过程中通过生产控制发现,随着目标产物的增长,一些次级代谢产物[如杂质A、B、C、D、E、F、G、H、I、FR1、FR2等(各杂质结构经核磁共振、液质联用确定,具体过程本文略,下同)]也会逐渐产生。这些次级代谢产物会降低目标产物的纯度,导致药品质量下降,再则这些杂质的毒理药理作用不明确,也可能影响临床药品的安全性、有效性。目前未见相关文献对非达霉素原料药在发酵产生过程中可能产生的杂质进行全面监控的报道。为了更好地控制非达霉素原料药质量,保证用药的安全性和有效性,笔者依据文献方法[16],对比研究了非达霉素原料药中主要杂质在正相-紫外检测器色谱系统、反相-蒸发光散射检测器色谱系统、反相-紫外檢测器色谱系统下的分离情况,建立以高效液相色谱(HPLC)法检查非达霉素原料药中有关物质的方法,并采用不加校正因子的主成分自身对照法计算杂质含量,为控制非达霉素原料药的质量提供参考。非达霉素的化学结构式见图1。
1 材料
1.1 仪器
2695型HPLC仪、2489型紫外检测器(美国Waters公司);2000ES型蒸发光散射检测器(美国Alltech公司);2010-CHT型HPLC仪(日本岛津公司)。
1.2 药品与试剂
非达霉素粗品(批号:20160601,纯度:90.2%)、非达霉素精粉(批号:20160801、20160802、20160803,纯度分别为:98.6%、98.8%、98.5%)、非达霉素对照品(批号:201510,纯度:98.8%)、非达霉素杂质C(批号:201607,纯度:98.6%)均由华北制药集团新药研究开发有限责任公司提供;乙腈、正己烷、乙醇均为色谱纯,其他试剂均为国产分析纯,水为自制蒸馏水。
2 方法与结果
2.1 色谱系统及检测器的选择
先后选用正相-紫外检测器色谱系统、反相-蒸发光散射检测器色谱系统、反相-紫外检测器色谱系统对非达霉素粗品进行分析,建立色谱条件。
2.1.1 正相-紫外检测器色谱系统检查有关物质
(1)色谱条件。色谱柱:ZORBAX Eclipse XDB-CN(250 mm×4.6 mm,5 ?m);流动相:正己烷-乙醇-乙腈(84 ∶ 13 ∶ 3,V/V/V,含0.1%醋酸和0.05%三乙胺);流速:1.5 mL/min;柱温:35 ℃;检测波长:245 nm;进样量:20 ?L。
(2)供试品溶液的制备。精密称取非达霉素粗品适量,加溶剂[乙醇-乙腈(5 ∶ 1,V/V)]溶解后,用正己烷定容并稀释制成每1 mL中约含2 mg非达霉素的溶液,即得。
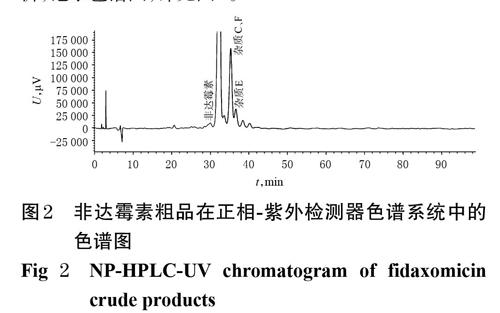
(3)非达霉素粗品分析。取“2.1.1(2)”项下非达霉素粗品供试品溶液,按“2.1.1(1)”项下色谱条件进样分析,记录色谱图,详见图2。
由图2可见,在本色谱条件下,色谱系统基线噪音大、信噪比高,杂质C与杂质F保留时间相同,分离较差;另外,还有部分含量在0.1%以下的杂质峰与噪音相当,不能检测出来。因此,正相-紫外检测器色谱系统不适用于非达霉素有关物质的检查。
2.1.2 反相-蒸发光散射检测器色谱系统检查有关物质
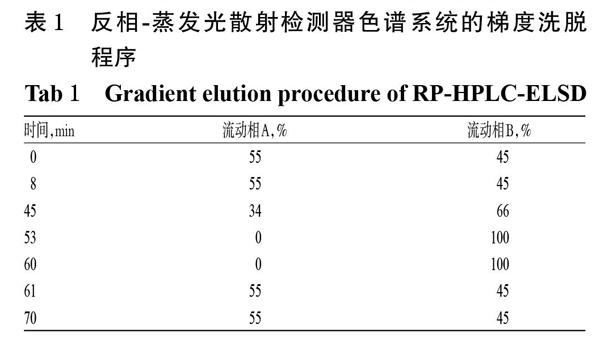
(1)色谱条件。色谱柱:Agilent Eclipse XDB C18(250 mm×4.6 mm,5 ?m);流动相:A相为0.05%冰醋酸缓冲液(用氨水调节pH至3.8)-乙腈(90 ∶ 10,V/V),B相为水-乙腈(10 ∶ 90,V/V),梯度洗脱;流速:1.0 mL/min;柱温:35 ℃;进样量:20 ?L;蒸发光散射检测器漂移管温度:110 ℃;气体流速:3.0 L/min。梯度洗脱程序见表1。
表1 反相-蒸发光散射检测器色谱系统的梯度洗脱程序
(2)供试品溶液的制备。精密称取非达霉素粗品适量,用乙腈定容并稀释制成每1 mL中约含5 mg非达霉素的溶液,即得。
(3)非达霉素粗品分析。取“2.1.2(2)”项下非达霉素粗品供试品溶液适量,按“2.1.2(1)”项下色谱条件进样分析,记录色谱图,详见图3。
由图3可见,非达霉素粗品在反相-蒸发光散射检测器色谱系统中仅能检出杂质C、D、E、F,其它杂质不能有效检出。因此,反相-蒸发光散射检测器色谱系统不适用于非达霉素有关物质的检查。
2.1.3 反相-紫外检测器色谱系统检查有关物质
(1)色谱条件。色谱柱:Agilent Eclipse XDB C18(250 mm×4.6 mm,5 ?m);流动相:A相为2‰三乙胺缓冲液(用磷酸调节pH至3.8)-乙腈(55 ∶ 45,V/V),B相为0.2%三乙胺缓冲液(用磷酸调节pH至3.8)-乙腈(20 ∶ 80,V/V),梯度洗脱;流速:1.0 mL/min;检测波长:230 nm;柱温:35 ℃;进样量:10 ?L。梯度洗脱程序见表2。
表2 反相-紫外检测器色谱系统的梯度洗脱程序
(2)供试品溶液的制备。精密称取非达霉素粗品适量,用乙腈定容并稀释制成每1 mL中约含0.5 mg非达霉素的溶液,即得。
(3)非达霉素粗品分析。取“2.1.3(3)”项下非达霉素粗品供试品溶液适量,按“2.1.3(1)”项下色谱条件进样分析,记录色谱图,详见图4。
由图4可见,非达霉素粗品在反相-紫外检测器色谱系统下,可检出非达霉素杂质A、B、C、D、E、F、G、H、I、FR1、FR2共计11个杂质,与前2种色谱系统比较,该色谱系统不仅检出杂质个数更多,而且杂质分离效果更好、灵敏度更高。因此,本研究选择反相-紫外检测器色谱系统对非达霉素有关物质进行检查。
2.2 有关物质检查方法的建立
2.2.1 溶液的制备
(1)系统适用性溶液。分别取非达霉素对照品及杂质C(用于考察其与主峰的分离情况)适量,用乙腈溶解稀释制成质量浓度均约为2.5 μg/mL的混合溶液,作为系统适用性溶液。
(2)供试品溶液。取非达霉素精粉适量,精密称定,加乙腈溶解并稀释制成每1 mL中约含0.5 mg非达霉素的溶液。
(3)对照溶液。精密量取“2.2.1(2)项下”供试品溶液1.0 mL,置于200 mL量瓶中,用乙腈稀释至刻度,摇匀,作为对照溶液。
2.2.2 专属性试验
(1)酸破坏样品溶液。取非达霉素精粉(批号:20160801,下同)25 mg,置于50 mL量瓶中,加1 mol/L 盐酸溶液5 mL,放置15 min后,加1 mol/L 氢氧化钠溶液5 mL中和,再加乙腈溶解,定容,摇匀,即得。
(2)碱破坏样品溶液。取非达霉素精粉25 mg,置于50 mL量瓶中,加0.01 mol/L 氢氧化钠溶液1 mL,放置15 min后,加0.01 mol/L 盐酸溶液1 mL中和,再加乙腈溶解,定容,摇匀,即得。
(3)高温破坏样品溶液。取非达霉素精粉适量,置于干燥坩锅中,放酒精灯上(外焰温度400~500 ℃)翻炒1 min左右,至颜色变淡黄后取出放冷,取25 mg,置于50 mL量瓶中,加乙腈溶解,定容,摇匀,即得。
(4)氧化破坏样品溶液。取非达霉素精粉25 mg,置于50 mL量瓶中,加30%过氧化氢溶液3 mL,于95 ℃水浴放置40 min,放冷,加乙腈溶解,定容,摇匀,即得。
(5)光照破坏样品溶液。取非达霉素精粉适量,置于强光照射试验箱[(4 500±500) lx]照射10 d后,取出放冷,取25 mg,置于50 mL量瓶中,加乙腈溶解,定容,摇匀,即得。
取“2.2.1(2)”项下供试品溶液和“2.2.2(1)~(5)”项下酸破坏、碱破坏、高温破坏、氧化破坏、光照破坏样品溶液各10 μL,按“2.1.3(1)”项下色谱条件进样测定,记录色谱图。结果,非达霉素精粉在各破坏条件下均有不同程度的降解,降解产物与主峰以及杂质峰间均能达到基线分离,提示本方法的专属性较强。6种溶液的色谱图见图5(注:图中RRT表示未知降解产物峰以主峰为基准的相对保留时间)。
2.2.3 检测限与定量限考察
将非达霉素精粉供试品溶液用乙腈适当稀释后,按“2.1.3(1)”项下色谱条件连续进样6次,记录峰面积。当信噪比为3 ∶ 1时得检测限;当信噪比为10 ∶ 1时得定量限。结果,非达霉素的检测限为0.05 ng,定量限为0.15 ng。
2.2.4 线性关系考察
取非达霉素对照品约50 mg,置于50 mL量瓶中,加乙腈溶解定容,然后逐级稀释为0.5、0.8、1.0、1.5、5.0、10.0、20.0 μg/mL的溶液,振蕩摇匀,按“2.1.3(1)”项下色谱条件进样测定,记录色谱图。以非达霉素质量浓度(x,μg/mL)为横坐标、峰面积(y)为纵坐标进行回归分析。结果,非达霉素回归方程为y=25 258.015 9x+238.201 9(R2=0.999 9),检测质量浓度线性范围为0.5~20.0 μg/mL。
2.2.5 重复性试验
精密称取非达霉素精粉约25 mg,平行称定6份,按“2.2.1(2)”项下方法制备供试品溶液,按“2.1.3(1)”项下色谱条件进样测定,记录峰面积,代入回归方程计算含量。结果,非达霉素含量的RSD为1.8%(n=6),表明该方法重复性良好。
2.2.6 中间精密度试验
由不同分析人员使用不同仪器,按“2.1.3(1)”项下色谱条件连续进样测定同一供试品溶液6次,记录峰面积,代入回归方程计算含量。结果,非达霉素含量的RSD为3.1%(n=6),表明该方法中间精密度良好。
2.2.7 回收率试验
精密称取非达霉素对照品9份,各约25 mg,置于50 mL量瓶中,用乙腈溶解并稀释至刻度,振荡摇匀,再分别稀释成0.1%、0.2%、0.3%浓度(非达霉素原料药的杂质限度是0.5%,但多批样品检测的杂质限度均在0.2%左右,故以0.2%作为中间浓度)的溶液各3份,作为供试品溶液。按“2.1.3(1)”项下色谱条件进样,记录色谱图,按外标法以峰面积计算回收率,结果见表3。
表3 回收率试验结果(n=9)
2.2.8 非达霉素有关物质检查
取非达霉素精粉3批,按“2.2.1(2)”“2.2.1(3)”项下方法制备供试品溶液和对照溶液,按“2.1.3(1)”项下色谱条件进样分析,记录峰面积,并按不加校正因子的主成分自身对照法计算各杂质(其中杂质C、E为已知结构的杂质)的含量。有关物质检查结果见表4。
表4 样品中有关物质检查结果(%)
3 讨论
3.1 色谱系统及检测器的选择
HPLC法为药物分析中供试品有关物质检查的常用方法,常用的有反相色谱系统和正相色谱系统。反相色谱系统使用非极性填充剂,常用洗脱剂为乙腈、甲醇等;正相色谱系统使用极性填充剂,常用洗脱剂为正己烷、异丙醇、无水乙醇等[17-18]。HPLC法中最常用的检测器为紫外检测器,而蒸发光散射检测器为通用型检测器,对所有化合物均有响应。
本研究比较了正相-紫外检测器色谱系统、反相-蒸发光散射检测器色谱系统、反相-紫外检测器色谱系统对非达霉素有关物质的检出能力,发现在采用反相-紫外检测器色谱系统检测时,其灵敏度高于反相-蒸发光散射检测器色谱系统,可检出的杂质种类更多,且非达霉素峰与各杂质峰的分离最好,专属性强、灵敏度高。因此,选择专属性更好、灵敏度更高的反相-紫外检测器色谱系统对非达霉素有关物质进行检查。
3.2 色谱条件选择
3.2.1 流动相pH选择
在反相-紫外检测器色谱系统中,笔者使用三乙胺、磷酸制备缓冲液调节pH,结果显示,当pH为4.5时,主峰后仅能分离出5个杂质峰,有1个杂质峰不能分离;当pH为3.0时,分离效果较好,但主峰后杂质峰的分离比略差,且色谱条件酸度较大,易损害色谱柱寿命;当pH为3.8时,分离情况好,主峰后各杂质峰均能有效分离,满足检查要求。因此,将pH为3.8定为本色谱条件下最佳pH条件。
3.2.2 流动相中三乙胺缓冲液浓度选择
在前期试验中,流动相中三乙胺缓冲液浓度分别选择了0.1%、0.2%和0.5%的3种浓度进行试验,结果发现浓度为0.2%时目标物质分离效果最好,出峰稳定且峰形尖锐,故选择三乙胺缓冲液浓度为0.2%。
综上所述,本研究建立的非达霉素原料药中有关物质的检查方法出峰快、峰形好、分离杂质多,且专属性好,适用于非达霉素原料药中的有关物质检查。
参考文献
[ 1 ] PARENTI F,PAGANI H,BERETTA G,et al. Lipiarmycin,a new antibiotic from actinoplanes:Ⅰ:description of the producer strain and fermentation studies[J]. J Antibiot,1975,28(4):247-252.
[ 2 ] JOHNSON AP,WILCOX MH. Fidaxomicin:a new option for the treatment of Clostridium difficile infection[J].J Antimicro Chem,2012,67(12):2788-2792.
[ 3 ] CAVALLERI B,ARNONE A,MODUGNO ED,et al. St- ructure and biological activity of lipiarmycin B[J]. J Antibiot,1988,3(3):308-315.
[ 4 ] ACKERMANN G,LOFFLER B,ADLER D,et al. In vitro activity of OPT-80 against Clostridium difficile[J]. Antimicrob Agents Chemother,2004,48(6):2280-2282.
[ 5 ] 姜春梅,刘洋,王京晶,等.抗感染新药非达霉素的药理作用与临床评价[J]. 中国新药杂志,2011,20(23):2283- 2285.
[ 6 ] 肖宇博,于峰.艰难梭菌感染的治疗新药:非达霉素[J].药学与临床研究,2012,5(22):419-424.
[ 7 ] 马培奇.美FDA咨询委员会推荐批准非达霉素治疗艰难梭菌感染[J].上海医药,2011,32(5):235.
[ 8 ] 黄仲义,黄嘉骅,赵永红.治疗难辨梭菌感染的新18元大环内酯抗生素非达霉素[J].上海医药,2014,35(1):50- 54.
[ 9 ] 闫宇青,李岩峰,高宇青.新型大環内酯抗生素非达霉素概述[J].药物流行病学杂志,2013,22(2):100-101.
[10] YE ZK,TANG HL, DUAN JL,et al. Efficacy and safety of fidaxomicin versus vancomycin for Clostridium difficile infection:systematic review and meta-analysis[J].J Chin Pharm Sci,2013,22(6):508-515.
[11] 路新华,郑智慧,丁彦博,等.一种游动放线菌菌株及其在制备非达霉素中的应用:中国 CN201310208223.2[P]. 2013-09-25.
[12] 高月麒,李晓露,王海燕,等.树脂吸附法分离纯化非达霉素的研究[J].中国抗生素杂志,2015,40(3):183-185.
[13] 李晓露,褚以文,王海燕,等.非达霉素活性降解产物的制备[J].中国抗生素杂志,2016,41(6):441-444.
[14] 刘月,王娅莉,任风芝,等.非达霉素潜在有机杂质的纯化与结构鉴定[J].中国抗生素杂志,2018,43(12):1498- 1501.
[15] 高月麒,任风芝,王海燕,等.非达霉素粗品中杂质的分离与鉴定[J].中国抗生素杂志,2015,40(11):828-830.
[16] 曹绪龙,祝仰文,严兰,等.氧化铝包裹硅胶核-壳型色谱填料的制备及正相色谱性能研究[J].分析实验室,2015,34(2):134-138.
[17] 韩真真,邵长森,张元元,等. HPLC法同时测定桂枝加芍药汤中8种成分的含量[J].中国药房,2019,30(6):784- 788.
[18] 陆文瑾,窦志华,曹瑞,等. HPLC法同时测定大黄药材中8个非蒽醌类成分的含量[J].中国药房,2019,30(14):1975-1980.
(收稿日期:2019-08-21 修回日期:2019-10-24)
(编辑:刘 萍)
