反相高效液相色谱法测定神经酸片剂中神经酸含量
2020-07-04吴乐艳侯雯清孙孔春杨璨瑜沈报春
吴乐艳,侯雯清,孙孔春,杨璨瑜,舒 纹,沈报春
(昆明医科大学药学院暨云南省天然药物药理重点实验室,云南昆明 650500)
神经酸(Nervonic acid) 因最早发现于哺乳动物的神经组织中而得名,其在周围神经组织和脑组织中含量较高,是神经组织中生物膜的重要组成成分,也是大脑发育及维持正常功能所必须的营养物质,对神经介质和受体发挥作用具有重要的意义[1]。有研究表明,神经酸具有增强体液免疫和细胞免疫[2]、改善记忆、认知功能损伤保护和抗氧化[3]作用,能疏通受损大脑神经通路并可促进神经细胞再生[4]。此外,神经酸对帕金森病[5-6]、脑白质疏松症[7-8]、Zellweger 综合征[9]、多发性硬化症[10]、肾上腺脑白质营养不良[11]、脑血管疾病[12-13]等有一定的治疗和预防作用。神经酸被认为是21 世纪最有前途的脑健康保健品和药品,为预防和治疗脑部疾病提供了一条新途径,具有广阔的应用前景[14]。
目前,神经酸定量分析多采用衍生化气相色谱法[15-18],该方法是将神经酸酯化后测定酯化物的含量,前处理过程较繁琐,且可能存在未反应的神经酸,导致神经酸含量测定不准确。文献[19]采用了高效液相色谱法测定蒜头果油分离出的神经酸含量,本文将建立一种反相高效液相色谱方法直接测定神经酸的含量,该方法不需对样品进行酯化,可直接进样分析,流动相组成简单,分析时间短,定量准确,可在短时间内实现样品的快速分析。
1 材料与方法
1.1 仪器与试剂
日本岛津LC-2010AHT 高效液相色谱仪(包含岛津SPD-M10A 检测器,岛津LC solution 色谱工作站);超纯水机(Direct-Q 3 UV):美国Millipore 公司;电子天平(BS224S):北京赛多利斯仪器系统有限公司;超声清洗仪(SK3200H):上海科导超声仪器有限公司;神经酸标准品:由云南盈木佳生物科技有限公司提供(纯度99.0%);神经酸片剂:某公司生产;乙腈(色谱纯):上海星可高纯溶剂有限公司;磷酸(分析纯)。实验用水为超纯水(电阻率为18.2 MΩ·cm,25℃)。
1.2 色谱分析条件
1.3 标准溶液配制
精密称取神经酸标准品0.050 4 g,以乙腈为溶剂,配制浓度约为2 mg/mL 的标准储备液。精密量取神经酸标准储备液1 mL 至10 mL 容量瓶中,加入9 mL 乙腈定容至刻度,摇匀,即得浓度为0.2 mg/mL 的标准溶液;再按照浓度梯度稀释法分别配制浓度为0.1 mg/mL、0.05 mg/mL、0.02 mg/mL、0.01 mg/mL 的多份标准溶液。
1.4 待测样品溶液配制
取神经酸片剂10 片,精密称定,研细。精密称取适量,置10 mL 容量瓶中,加乙腈适量,超声处理使主成分溶解,乙腈定容至刻度,充分摇匀,得待测样品溶液,备用。
1.5 测定
将标准溶液及待测样品溶液先用0.22 μm 的有机系滤膜过滤,取续滤液以10 μL 的进样量上样于高效液相色谱仪,记录保留时间、峰面积等参数。按外标标准曲线法以峰面积计算,即可测得神经酸片剂中神经酸的含量。
2 结果
2.1 方法学验证
2.1.1 标准曲线精密吸取神经酸系列标准溶液各10 μL,照“1.2.1”项下色谱条件进样分析,以各份标准品溶液的浓度(X,mg/mL) 为横坐标,以神经酸色谱峰的峰面积(Y) 为纵坐标作图(见图1),得线性回归方程y=3 000 000 X(mg/mL)+4 795.9,R2=0.999 9。神经酸含量在0 .01~0.2 mg/mL 浓度范围内具有良好的线性关系(P=0.000)。
2.1.2 精密度实验精密吸取2 mg/mL 神经酸标准储备液10 μL,照“1.2.1”项下色谱条件,连续进样6 次,记录峰面积,结果见表1。峰面积相对标准偏差RSD 为1.80%,说明实验方法精密度良好。
2.1.3 稳定性实验精密吸取待测样品溶液10 μL,照“1.2.1”项下色谱条件,在24 h 内测定6次(0 h、2 h、4 h、8 h、12 h、24 h),记录峰面积,结果见表1。由表1可见,峰面积相对标准偏差RSD为1.52%,待测样品溶液在24 h内稳定性良好。
2.1.4 加样回收率照“1.2.1”项下色谱条件,首先测定待测样品溶液中神经酸的峰面积,带入“2.1.1”项下标准曲线,求得待测样品溶液的浓度。接着将待测样品溶液平均分成9 份,再分成3 组,每组分别添加相当于待测样品溶液中神经酸含量80%、100%、120%三个水平的神经酸标准溶液,混匀后照“1.2.1”项下色谱条件分析,结果见表2。由表2 可见,添加神经酸标准溶液后所得样品的回收率在96.94%~104.46%之间,峰面积相对标准偏差RSD 在0.81%~3.80%之间,平均回收率为101.53%。该方法准确度良好,可用于神经酸片剂中神经酸的定量分析。
2.2 样品含量测定

图1 神经酸线性回归方程Fig.1 Linear regression equation of nervonic acid

表1 精密度实验、稳定性实验结果(1)Tab.1 Precision and stability results (1)

表1 精密度实验、稳定性实验结果(2)Tab.1 Precision and stability results (2)

表2 加样回收率实验结果Tab.2 Spiked recoveries and RSDs results

图2 标准品和样品典型色谱图Fig.2 Representative chromatograms of standard and sample
按照“1.2.3”项下方法,分别精密称取约相当于神经酸10 mg 的片粉,制备两份神经酸样品溶液,采用建立的高效液相色谱方法测定两份样品溶液中神经酸的含量,典型色谱图见图2,测定结果见表3。
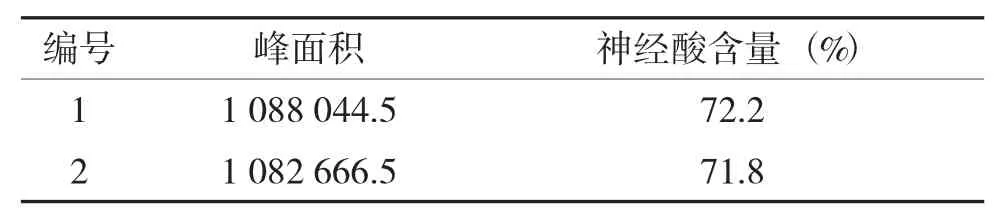
表3 样品测定结果Tab.3 Analytical results for the sample
3 讨论
神经酸不仅可以修复受损的神经纤维,而且还可促进受损的神经细胞再生,对动脉粥样硬化、高血脂、脑梗塞、脑出血、脑萎缩、痴呆、小儿脑瘫、癫痫、帕金森病、脑外伤等疾病的防治有一定作用。国外对神经酸的研究已开展了将近一个世纪,但在我国尚处于起步阶段,对于其来源、获取方法、药理作用、生理作用及新剂型、应用等的研究还有很大空间。
本课题组在前期实验中曾采用现有报道的衍生化气相色谱法测定神经酸片剂中神经酸含量,先将样品进行脂肪酸甲酯化,再用有机溶剂进行提取,气相色谱法测定,外标法定量。实验过程中发现由于神经酸沸点较高,采用将神经酸进行甲酯化后再定量的方法,可能存在神经酸甲酯化不完全的问题;甲酯化后又经过正己烷提取的操作,整个含量测定过程前处理操作步骤较多,会存在神经酸损失导致测定值比真实值偏小、定量不准确的问题,提取回收率较低;前处理过程复杂也存在方法的重复性及可操作性不强等缺陷。基于此,笔者建立了一种直接测定样品中神经酸含量的反相高效液相色谱方法,提高了神经酸定量的准确度。
本研究建立的含量测定方法,首次采用C8柱,神经酸出峰时间大概在20 min,具有分析时间短,分析效率高等优点[20],实现了对神经酸片剂中神经酸含量的准确测定,该方法不需要进行衍生化,前处理过程简单易操作,分析条件温和,分析速度快,定量结果准确。此外,该方法的色谱条件、流动相组成简单,精密度、重复性及RSD符合相关规定,稳定性好,灵敏度高,选择性好。可实现人力、物力、财力的节省,有利于神经酸资源的开发及运用,具有较好的应用前景。
