SLC4A1 复合杂合突变致遗传性球形红细胞增多症并远端肾小管酸中毒1 例报告并文献复习
2020-06-30许珊珊白海涛
陈 雁 许珊珊 白海涛 杨 阳
厦门大学附属第一医院儿科 厦门市儿科重点实验室 厦门大学医学院儿童医学研究所(福建厦门 361000)
由人类SLC4A1基因编码合成的阴离子-共同转运体(anion exchanger1,AE1)是重要的碳酸氢盐转运体。AE1的红系亚型(erythroid anion exchanger 1,eAE 1)是红细胞中最丰富的蛋白质,其羧基端的跨膜结构域是红细胞骨架蛋白的锚定位点,氨基端负责细胞内外Cl-/HCO3-离子转运。同时AE 1 又以缺失氨基端65个氨基酸残基的截短形式(truncated kidney isoform of Anion exchanger1,kAE1)表达于肾脏A型闰细胞(α-intercalated cells,A-ICs),主要负责HCO3-分泌入血同时将Cl-重吸收[1-2]。发生在AE 1 编码区域内的突变都可能导致红细胞形态异常并发生溶血性贫血或者远端肾小管酸中毒,只有极少部分表现为两者并存[3]。现回顾分析1例罕见的由于SLC4A1基因复合杂合突变导致的遗传性球形红细胞增多症(hereditary spherocytosis,HS)合并远端肾小管酸中毒(distal renal tubular acidosis,dRTA)患儿的临床资料,并复习相关文献。
1 临床资料
患儿,男,1 岁7 个月。患儿生后3 天因皮肤黄染2 天入住当地医院,考虑新生儿高胆红素血症,光照治疗5 天,皮肤黄染消退后出院。出院时家属即感患儿面色口唇欠红润,添加辅食后仍无改善,且患儿活动少。8 月龄时外院就诊,骨髓穿刺检查示可见球形红细胞,骨髓红系增生活跃,巨核细胞增多。考虑诊断HS,定期输血治疗,乏力症状于输血后改善。本次入院前2 天患儿再次出现食欲减退、乏力、活动减少,间断呕吐胃内容物,来厦门大学附属第一医院就诊。患儿父母非近亲结婚,否认相关疾病家族史。入院体格检查:体温36.8℃,呼吸26次/min,心率118次/min,血氧饱和度98%,体质量9 kg(<P3),身高70 cm(<P3);神清,倦怠,贫血貌,巩膜及皮肤轻、中度黄染,独走不稳,双肺呼吸音粗,腹部稍胀,肝肋下3~4 cm,质软,脾肋下3 cm,质中;腱反射弱。实验室检查:血常规白细胞10.48×109/L,红细胞2.15×1012/L,血小板283×109/L,血红蛋白67 g/L,MCV 84fL,MCHC34.6g/dL,网织红细胞2.9%;血气分析pH 7.25,PCO23.27 kPa,PO217.9 kPa,剩余碱-15.4 mmol/L;血钠134 mmol/L,钾2.7mmol/L,氯113 mmol/L,CO28.5 mmol/L,钙2.08 mmol/L,肌酐40 μmol/L,丙氨酸转氨酶 15 U/L,天冬氨酸转氨酶 22 U/L,总胆红素 42 μmol/L,间接胆红素 40 μmol/L,碱性磷酸酶489μmol/L;直接、间接抗人球蛋白试验均阴性;红细胞渗透脆性试验:开始溶血0.6%,完全溶血 0.37%(均按照氯化钠溶液浓度计算);尿常规pH 7.0,尿比重 1.008,余未见异常。泌尿系B 超及心脏彩超未见异常。入院诊断:HS,代谢性酸中毒,电解质紊乱(高氯血症及低钾血症)。
患儿入院后经碳酸氢钠输注及口服枸橼酸钾纠正酸中毒及低钾血症、输注红细胞纠正贫血等处理,病情好转。为明确病因,经医学伦理审核、患儿父母签署知情同意书后采集患儿及父母外周血样各2 mL,EDTA 抗凝后送检全外显子基因组测序,并经一代测序验证。检出SLC4A1基因两个等位基因变异,等位基因c.2102G>A(p.Gly701Asp)来源于母亲,为已知的致病性变异[1];等位基因c.1988T>C(p.Met663Thr)来源于父亲,为已知的与HS 相关的变异。见图1。患儿治疗1 周后出院,目前仍门诊随访,定期复查血常规、电解质及输血治疗。
2 讨论
SLC4A1是SLC4家族成员之一,定位于17号染色体q21~q22。由其编码的eAE1亚型表达于红细胞胞膜,又名带-3蛋白(Band-3,B3),维持红细胞正常骨架结构及离子传输,因此SLC4A1突变可能导致红细胞形态异常,临床表现包括HS、东南亚卵形红细胞增多症、遗传性口型红细胞增多症及遗传性干瘪红细胞增多症等疾病 。kAE1亚型表达于肾脏闰细胞侧基膜,主要负责阴离子转运,SLC4A1的突变导致尿液离子转运障碍,尿液不能正常酸化,出现dRTA[3]。
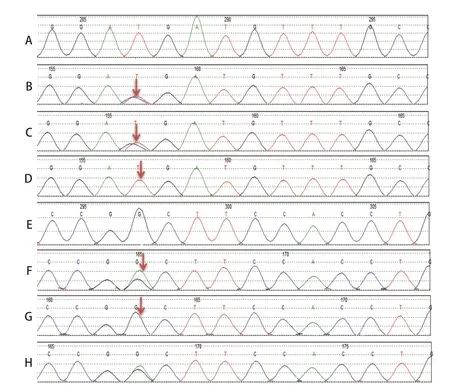
图1 患儿及父母SLC4A1 基因Sanger 测序峰图
多数SLC4A1突变仅导致dRTA或红细胞形态异常,只有少部分突变二者同时存在。这种情况也发生在基因敲除小鼠以及人类纯合子HS 突变中[4]。体外研究发现,发生在AE1基因编码序列外显子区域的错义、无义和移码突变都可能导致eAK 1 氨基酸残基改变,患者出现红细胞形态异常,但未必合并dRTA。这是因为,红细胞以糖蛋白A(glycophorin A,GPA)的形式表达了一个eAE 1 伴侣样分子,GPA可以携带突变蛋白逃逸出内质网的俘获到达细胞膜表面,参与离子转运,挽救大多数导致dRTA变异[5]。而包括G701D、S773P及ΔV850在内的位点突变可影响kAE1中央跨膜结构域和短羧基端尾部,使GPA 发生错误折叠,不能经正常途径分泌至细胞质膜参与离子转运,只能保留在内质网或高尔基体中最终被溶酶体靶向降解,患者出现尿液酸化障碍[6]。换而言之,患者的临床症状取决于突变的eAE1是否能与野生型蛋白(wild type protein)形成异二聚体,以及该突变蛋白与GPA的互动能力。
SLC4A1基因突变可有常染色体显性及常染色体隐性两种遗传方式,在全球不同地区呈现不同的临床表现和遗传模式[3],从东南亚地区至印度洋、地中海沿岸都有分布。已报道的常染色体显性遗传突变位点11个,常染色体隐性遗传突变位点18个[3-14](表1)。常染色体显性遗传-AE1 少见且症状隐匿,主要出现在温带国家,患者总体起病年龄较晚,症状缺乏典型性,往往以低钾血症导致麻痹、乏力及骨质疏松生长迟缓为首发症状就诊;常染色体隐性遗传-AE 1 报道较多,主要出现在热带国家,尤其以东南亚地区最为常见,临床表现差异较大但总体症状较严重。G701D是目前报道病例最多的突变位点[7~9]。研究报道5/8的泰国人呈现G701D 纯合突变,却仅有红细胞轻度形态异常而无溶血性贫血表现[10],说明AE1 G701D是泰国常见的分子缺陷。菲律宾对7 个dRTA 的家系成员研究发现均符合G701D纯合突变或复合杂合突变[11]。SAO 是另一个目前报道较多的由AE1(ALA400-ALA408)突变引起的疾病,目前仅见于东南亚及巴布亚新几内亚等热带地区,导致卵母细胞红细胞产生抗药性的突变以防止疟原虫侵袭。曾有涵盖9个家系13例患者的病例报告,年龄5个月~ 36岁不等,其中3个家系来自马来西亚,均为常染色体隐性遗传,2个家系突变位点为G701D/SAO,1家系为A858D/SAO,6 个家系来自巴布亚新几内亚,其中5 个家系为常染色体隐性遗传,1例突变位点分别为ΔV850/ΔV850,4例为ΔV850/SAO,仅1例常染色体显性遗传,突变位点为ΔV850/A858D。13例中11例初诊时呈现完全型远端肾小管酸中毒,9例有肾钙化/结石形成,8例初诊时血钾<3.0 mmol/L,最低为1.6 mmol/L,而只有4例患者初诊时血红蛋白<90 g/L[12]。曾报道1例葡萄牙籍患儿,胎儿期因严重溶血性贫血及胎儿水肿综合征导致宫内窘迫,紧急剖宫产并经抢救及输血后存活,生后反复多次输血并定期去铁治疗;3 月龄时发现合并低钾高氯酸中毒及肾钙化,口服碳酸氢钠及磷酸钾纠正,9 月龄行脾切除术;随后证实患儿系V488M 纯合突变,导致AE 1 第4 个跨膜结构域起始处发生异常[13]。这是目前报道的起病最早的患儿。

表1 不同遗传方式下SLC4A1基因突变比较
近年来AE1复合杂合突变导致dRTA并溶血性贫血的个案也陆续报道。2009年中国台湾报道1例7月龄男婴,以严重溶血性贫血及dRTA起病,首诊时血红蛋白42 g/L,Sanger测序证实其携带 E522K/G701D复合杂合突变,而基因型为E522K/WT 的同胞哥哥及母亲仅表现为轻度的红细胞形态异常而无贫血及dRTA症状,基因型为G701D/WT的父亲无红细胞形态异常及电解质紊乱表现[4]。进一步的体外实验证实,kAE1WT 蛋白可以协助突变的kAE1G701D 转移到细胞膜表面参与离子交换,而kAE1E522K 缺乏相应功能。有报道1例携带C479W/G701D复合杂合突变的患者,红细胞表达AE 1 显著减少,经体外实验发现该突变蛋白虽然能够形成异二聚体,但彼此独立流动,导致碳酸氢盐无法重新吸收到血液中,推测C479W 突变体是一个新的AE 1 转运突变体,由于细胞表面AE 1 蛋白降低而导致HS,并由于其在肾脏闰细胞内滞留而导致dRTA[14]。此外,转染细胞实验证实G494S/G701D、S773P/G701D等复合杂合突变也可导致红细胞包膜异常并离子转运功能丧失[3,15]。
M663T突变目前仅报道1例,患者初诊年龄17岁,流式细胞学检测提示正常eAK1含量54%,其父母及表型正常的兄弟姐妹eAK1含量为72%~124%。进一步研究发现,该变异使得红细胞胞膜疏水氨基酸转变为亲水氨基酸导致胞膜结构异常而未影响GPA功能,使得闰细胞内离子转运得到代偿,从而解释患者仅有轻度溶血性贫血而无肾小管酸中毒临床表现[16]。
本例患儿起病年龄小,新生儿期即出现溶血性贫血表现,8月龄开始定期输血支持,低钾血症导致的乏力症状在每次输血后得以缓解,一定程度上也掩盖了病情。初诊时辅助检查结果呈现经典的dRTA,符合常染色体显性遗传临床表现的特点,即起病早、症状重、临床表现复杂及多系统受累。因患儿年龄较小,故目前尚未出现听力异常及泌尿系结石改变,有待远期进一步随访。
伴溶血性贫血的肾小管酸中毒治疗目标是纠正代谢性酸中毒及定期输血支持,同时避免并发症出现,包括生长发育迟缓、佝偻病及肾结石,注意祛铁治疗,年长儿可考虑脾切除术甚至骨髓移植[17]。需要关注的是肾钙盐沉积,可适当增加膳食中果汁与柑橘类水果比例,补充更多的柠檬酸有利于肾结石排出,正常补钙,并限制草酸盐、动物蛋白等摄入,尿中低分子蛋白水平是患者治疗依从性或充分性良好的标志[18]。治疗期间需要定期评估超声,确保肾结石/钙化形成时及早发现。肾小管酸中毒预后与是否早期充分纠正酸中毒、纠正低钾血症有关,远期约有1/3 患者仍将进入慢性肾脏病阶段。
