CL-RBF:一种基于改进ML-RBF 的蛋白质亚细胞多点定位预测算法
2020-05-11洪晓宇胡雪娇陈行健
薛 卫, 洪晓宇, 胡雪娇, 陈行健, 张 梁
(1. 南京农业大学 信息科学技术学院,江苏 南京210095;2. 江南大学 粮食发酵工艺与技术国家工程实验室,江苏 无锡214122)
蛋白质是生命体内功能最丰富的生物大分子,在必需的生命活动中,发挥着关键性作用。 其功能的正常发挥依赖其所在的亚细胞位置,只有处于特定的细胞器上,蛋白质才能正常发挥作用,从而保证生命体的正常运转。 因此蛋白质亚细胞定位预测在识别未知功能的蛋白质序列和确定基因组标注中都有重要的意义和作用,还可以极大地提高药物靶点的识别[1]。 随着生物数据快速更新和大量积累,使用人工实验去获取蛋白质的位置已远不能达到科研需要,从而促使了机器学习在蛋白质亚细胞定位预测中的发展。 1991 年,Nakai 和Kanehisa[2]在对革兰氏阴性菌蛋白质进行亚细胞位置识别时,整理出了一系列选择判别规则,将机器学习的方法首次应用到蛋白质亚细胞定位预测中。 2000 年,Cai 和Chou[3]采用了基于人工神经网络的自组织模型来预测原核和真核生物蛋白质的亚细胞位置。 2013 年,曹隽喆[4]建立了一种不平衡权重的多标签尺KNN模型,以消除蛋白质数据集分布不均衡的情况。 Yang等人[5]在2014 年提出了一种基于可能性的SVM 模型被用来识别人类蛋白质亚细胞位置。 2015 年,Liu和Tao 等人[6]将基于最大间隔原理的SVM-RFE 算法与基于PSSM (Position-Specific Score Matrix)的Tri-gram 编码方式结合, 在3 个不同的数据集上都取得了较好的实验结果。 Bendtsen 等人[7]在SignalP方法的基础上进行了改进, 提出了SignalP 3.0.Rahman 等人[8]将蛋白质的多种特征融合,提出了AAIDPAAC 和PPMPAAC 特征,与SVM 结合得到了较高的预测准确率。
在人工神经网络应用的早期,BP 神经网络、概率神经网络和SOM 使用较多, 但后来逐渐被径向基(Radial Basis Function, RBF)神经网络所取代[9]。为了解决多目标分类问题,Zhang[10]提出了一种基于RBF 神经网络的多目标学习方法ML-RBF。 该算法的训练一般分为两步: 第一步在不同的目标下,对属于该目标的训练样本进行K-means 聚类得到隐层中心,将原型向量与隐层中心关联,计算径向基函数;第二步通过最小化误差平方和来获得最优化权重。 因此对ML-RBF 算法的改进也主要集中在:结构设计,即隐藏层节点设计;参数优化,包括基函数的数据中心及扩展常数、输出节点的权值等模型参数。 作者分别从这两点出发并结合实验对象特点对ML-RBF 算法进行改进,提出CL-RBF 算法。 实验结果显示,CL-RBF 算法在不同的数据集上都显示出了较高的性能。
1 材料与方法
1.1 数据集
Xiao 等人[11]指出细菌蛋白质在生命体活动中是一把”双刃剑”,既存在巨大危害,又发挥着不可忽视的积极作用,因此被认为是最有研究价值的蛋白质之一,故选取细菌作为实验对象。 目前数据集构建方法可分为3 种: 一是直接选用已有数据集;二是选取已有数据集并进行更新;三是重新构建数据集。 直接用前人提出的数据集进行实验的弊端是忽略了时间间隔内的数据更新问题,因此选取后两种方式进行数据集构建。原始数据集都来自于UniProt数据库,通过CC(comment or notes)和OC (organism classification)检索框来搜索革兰氏阴性菌和革兰氏阳性菌的蛋白质序列。 所有数据必须严格按照下列标准进行筛选:
1) 革兰氏阴性菌: 在OC 检索框中输入“proteobacteria”;革兰氏阳性菌:在OC 检索框中输入“firmicutes” 和“actinobacteria”。
2) 在CC 检索框中选取subcellular location term,由于实验针对的是多目标,因此不选取具体的亚区间,以* 代替,蛋白质序列只选取经过实验验证的,即“experimental assertion”。
3) 蛋白质序列的长度规定在50~10 000 区间,并且不能是片段。
通过筛选后得到原始的蛋白质序列集合,由于过高的相似度会增加实验的准确率,为了使训练出的模型更加客观, 利用CD-HIT 设置相似度阈值为30%,剔除相似度过高的蛋白质序列。再采用网络爬虫技术到UniProt 数据库中抓取亚细胞信息, 具体抓取条件如下:
1)尽管表述方式不同,但是多个关键字可能代表同个细胞区域。关键字“extracell”, “extracellular” 和“secreted” 可以相互替换; 对于革兰氏阴性菌,“cytoplasm”和关键字“cytoplasmic”具有同样的意义。 对于革兰氏阳性菌,当检索蛋白质亚细胞时,关键 字 “plasma membrane”, “integral membrane”,“multi-pass membrane”和“single-pass membrane”都归类于亚细胞“cell membrane”。
2)当蛋白质序列被模糊或者不确定项标注时,例如“potential”, “probable”, “probably”, “maybe”,或者“by similarity”则会被剔除。
经过处理后得到革兰氏阴性菌数据集革兰氏阴性菌833 和革兰氏阳性菌数据集革兰氏阳性菌464[12-13]。 为了维持样本的均衡性,选择革兰氏阴性菌和革兰氏阳性菌蛋白质序列分布较广的亚细胞[14]。革兰氏阴性菌833 数据集包含5 个亚细胞,分别是细胞内膜 (Cell inner membrane)、 细胞外膜(Cell outer membrane)、细胞质(Cytoplasm)、细胞外基质(Extracellular)和细胞周质(Periplasm);革兰氏阳性菌464 数据集包括细胞膜(Cell membrane)、细胞壁(Cell wall)、细胞质(Cytoplasm)和细胞外基质(Extracellular)4 个亚细胞, 数据集的具体分布情况见表1-2。
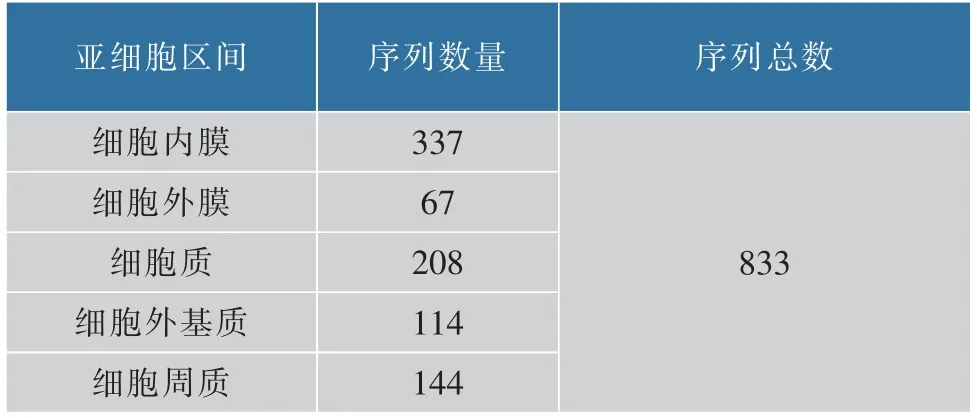
表1 革兰氏阴性菌833 数据集构成Table 1 Distribution of gram-negative833 dataset
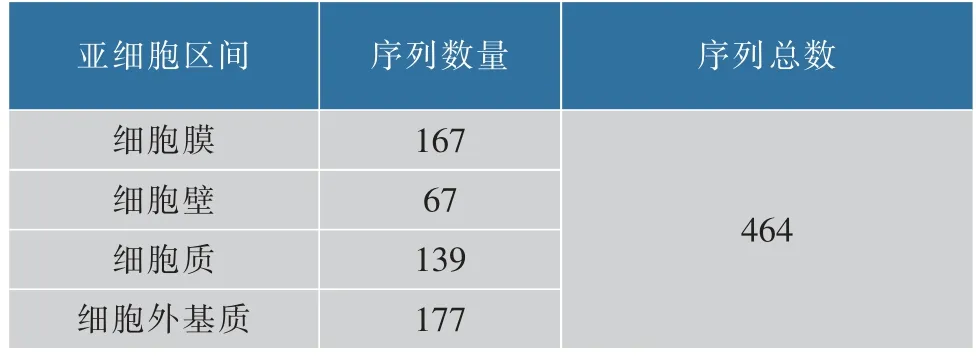
表2 革兰氏阳性菌464 数据集构成Table 2 Distribution of gram-positive464 dataset
此外,作者还对两个已经构建好的革兰氏阴性菌数据集[15]和革兰氏阳性菌数据集[16]进行筛选和重构。剔除革兰氏阴性菌中样本数量较少的亚细胞,即菌毛(Fimbrium),鞭毛(Flagellum)和拟核(Nucleoid)。利用网络爬虫技术重新获取亚细胞信息,从而使更新后的数据集能反应最新的序列信息。 亚细胞抓取和处理过程同数据集革兰氏阴性菌833 和革兰氏阳性菌464。 重构后的数据集分别为革兰氏阴性菌1392 和革兰氏阳性菌519,数据集的具体分布见表3-4。
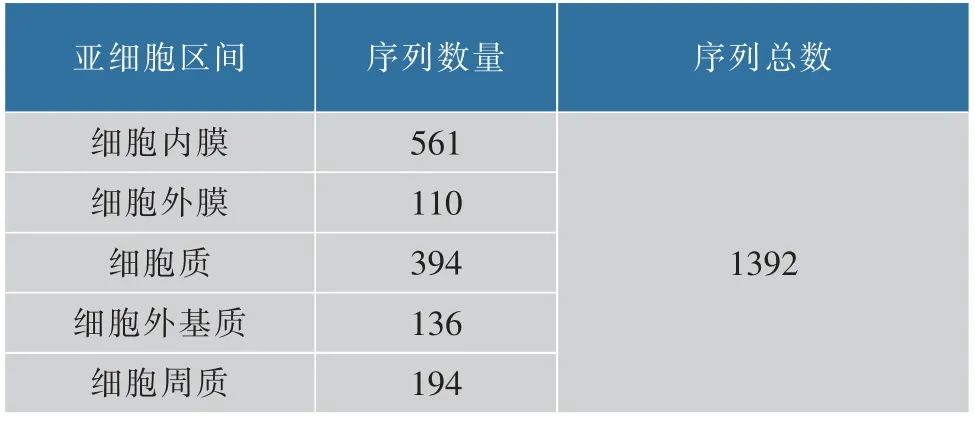
表3 革兰氏阴性菌1392 数据集构成Table 3 Distribution of gram-negative1392 dataset
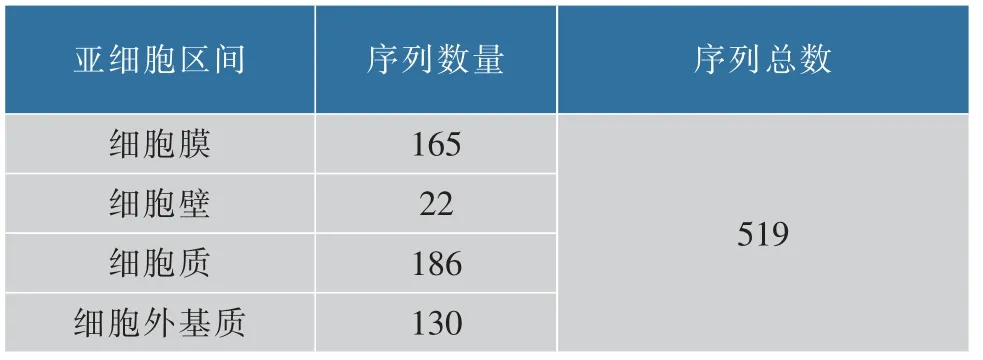
表4 革兰氏阳性菌519 数据集构成Table 4 Distribution of gram-positive519 dataset
1.2 序列特征编码
对蛋白质序列进行特征提取是蛋白质亚细胞定位预测中的重要环节,有效的特征提取方法可以增加预测准确率。 赵南等人[17]将词袋模型应用到蛋白质特征提取当中,并与传统的基于氨基酸组成的序列表示方法相结合, 提出了词袋特征这个概念。实验证明,词袋特征能有效增加分类器的预测准确率。 因此本研究采用词袋模型结合AAC 方法表示一条蛋白质序列。
1.2.1 氨基酸组成法 Nakashima 和Nishikawa[18]最早将氨基酸的组成和蛋白质亚细胞位置联系起来,提出AAC 编码方式, 统计每个氨基酸在蛋白质序列中出现的频率。
AAC 方法定义如公式(1)所示:
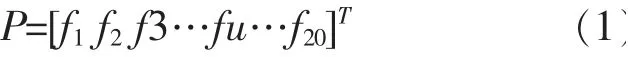
fu(u=1,2,3,…,20)为氨基酸u在序列中出现的频率;fu的求解方法如公式(2)所示:

其中,L表示一条蛋白质序列的长度,即包含的所有氨基酸残基的总数目。 首先要对20 种氨基酸从1到20 进行编号。
1.2.2 词袋特征 通过AAC 算法计算蛋白质序列P 的序列单词特征,可得一个片段特征矩阵,如公式(3)所示:
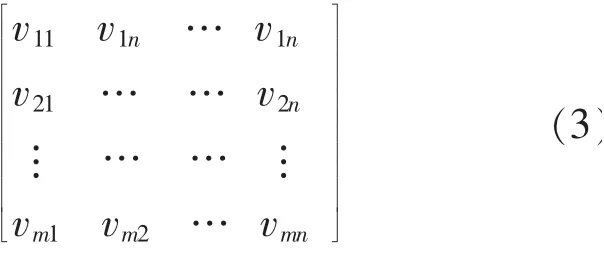
式(3)中,v表示蛋白质片段中氨基酸出现的频率;m表示一条蛋白质序列切割成的片段条数;n为经过特征提取算法处理后的特征维度。 具体的求解方法见文献[17]。
通过滑动窗口分割法和基于类内方差和最小的聚类方法得出一个最优的词袋字典, 字典包含i种不同的类别,每一类都是一个n维的向量,具体如公式(4)所示:

式(4) 中,di1di2din表示第i类的词袋特征,式(5)为词袋特征中第q维的计算方法:
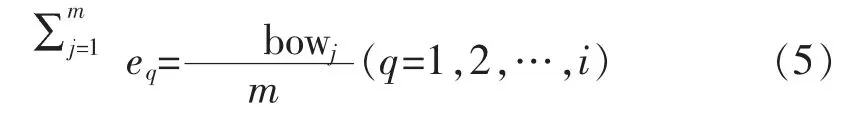
其中,bowj表示片段j是否属于字典中的q类。具体解决方法如下:

通过计算欧氏距离判断片段是否属于第q类,如果片段j与q类的欧式距离最小,则片段j属于q类。 最后得到完整的词袋特征,如公式(6)所示:

1.3 基于改进ML-RBF 的分类预测算法
1.3.1 基于标记内的聚类方法改进策略 聚类是一种无监督的学习方法,理想的聚类结果一般是类内间距最小,类间间距最大。 Kaufman 等人[19]基于上述思想,提出了轮廓系数S以及个体轮廓系数si,具体表述方式如下:
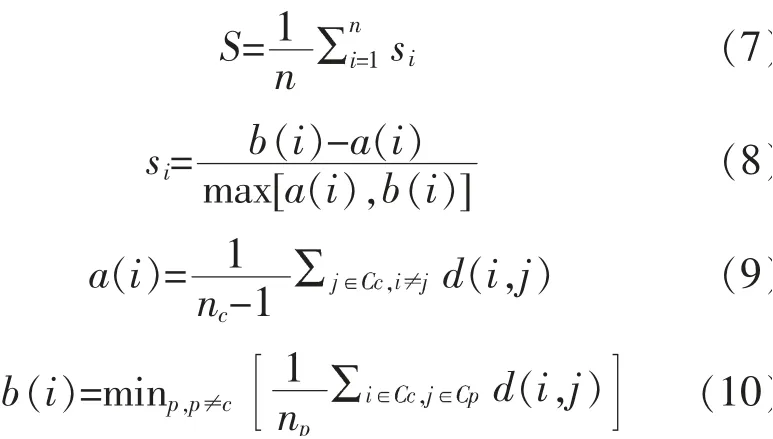
其中,n表示样本个数。 假设样本i属于聚簇c,a(i)表示样本i和同属于聚簇c的其他样本距离的平均值。b(i)选取样本i和不属于聚簇c的每个聚簇中所有样本的平均距离的最小值。
ML-RBF 神经网络的预测效果与隐层中心有密切的关系, 依次对每个类别标记, 采用传统Kmeans 算法对所属样本进行聚类, 计算不同聚类中心个数下的轮廓系数。 轮廓系数越大,说明对应类别标记选取的聚类中心个数越合理。 具体的改进步骤如下:
1)对于每一个l∈L,设置聚类个数k的搜索范围为k∈[kmin,kmax]。
2)forkmintokmax。
●调用传统K-means 算法对属于类别标记l的样本进行聚类;
●利用公式(8)计算个体轮廓系数si。
●利用公式(7)计算轮廓系数S。
3)选取最大轮廓系数对应的聚类个数,作为类别标记l的最佳聚类个数kbest。
1.3.2 基于标记间的聚类方法改进策略 以往对ML-RBF 中隐层中心的改进主要针对同一类别标记内的聚类结果进行优化,从而忽略了不同类别标记间的隐层中心之间的相互影响。 作者提出了一种基于类别标记间的聚类方法改进策略。 该方法将标记间(Inter-Label)隐层中心距离与一个预设的阈值γ 进行对比,从而调整不同聚簇中的样本,进而重新计算聚簇中心(隐层中心)。
阈值γ 的公式如下:

其中,α 为一个常数;μ 为比例因子。
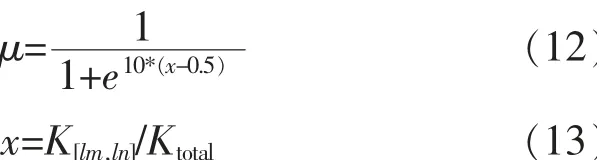
当x很小时,比例因子的下降速度很慢,比例因子接近于1,阈值γ 无限接近于α。
改进的具体流程如下:
1)计算不同类别标记间的隐层中心距离dist。
2)将隐层中心之间的距离从小到大排序,并生成隐层中心对集合Dtwo={(cip,cjq)|dist(cip,cjq)<α}p,q∈L
3)从集合Dtwo选取一对隐层中心(cilm,cjln)进行调整,同时从集合Dtwo将其删除。
4)利用公式(11)~(13)计算阈值γ。
5)for dist(cilm,cjln)<γ
6)直到Dtwo=Ø,算法结束,否则跳转到3)。
1.3.3 自适应梯度下降调整参数 为了进一步缩小误差,使用梯度下降的学习方法对平滑指数σi以及隐藏层与输出层的链接权重W进行参数调整。由于求解权重W产生的误差较大, 初始学习率设置η1>η2,η1为调节权重的学习率,η2为调节平滑指数的学习率。 通过在革兰氏阳性菌和革兰氏阴性菌数据集上反复试验,发现两者采用不同步的调整策略能更快地逼近最小值。 当代价函数增大时,减小权重步长;当代价函数减小时,增加平滑指数步长。
1.3.4 基于聚类优化的结果集调整策略X=Rd为输入空间,给定一个多标记训练集合D={(xi,Yi)|1≤i≤m},其中xi∈X表示一个训练示例,Yi⊆L表示与示例xi关联的类别标记集合。
针对某个类别标记l, 分别有Xpositive={xi|(xi,Yi)∈D,l∈Yi},xi∈X表示训练样本xi属于类别标记l,该训练样本构成集合Xpositive;Xnegative={xj |(xj,Yj)∈D,l∉Yj},xj∈X表示训练样本xj不属于类别标记l,该训练样本构成集合Xnegative。并且有Xpositive∩Xnegative=Ø,Xpositive∪Xnegative=X。
在传统的ML-RBF 训练中,第一步仅考虑对集合Xpositive进行聚类,而忽略了对Xnegative进行聚类,分析两者的关系。 对Xpositive和Xnegative两种样本分别进行聚类, 当待测样本与Xpositive的聚类中心距离更近时,说明测试样本与l类的样本Xpositive相似度更高,预测值应该增大;反之应该减小。 对Xpositive通过基于标记内和标记间改进的K-means 聚类得到kpositive个隐层中心,构成集合对Xnegative同样采用引进个体轮廓系数的方法进行聚类,生成knegative个聚类中心,构成集合在调整了类别标记间隐层中心距离的前提下,当测试样本x与隐层中心cip的距离dist(x,cip)越小,则x越可能被认为属于类别标记l, 然而此时若有dist(x,cjn)<dist(x,cip),则说明x与不能归类于类别标记l的训练样本的聚簇中心更近;反之亦然。调整过程如下:
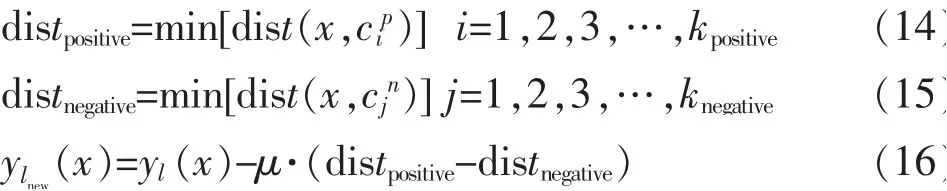
distpositive为x到Cpositive中每一个中心的最小距离,distnegative为x到Cnegative中每一个中心的最小距离。 μ 为比例因子, 本研究设置为1, 当distpositive<distnegative,ylnew(x)输出结果增大,反之减小。
1.3.5 基于CL-RBF 的蛋白质亚细胞定位预测器将CL-RBF 应用到蛋白质亚细胞定位预测中,具体流程如下:
1)计算数据集中所有蛋白质序列的词袋特征,把词袋特征集合作为样本集合。 将所有序列的词袋特征分为n组互不相交的子集合,依次取出一个子集作为测试集,其余n-1 个子集构成训练集。
2)针对某个亚细胞i,将训练样本分为属于亚细胞i的训练样本和不属于亚细胞i的训练样本,分别对两种训练样本使用基于标记内改进的Kmeans算法进行聚类, 得到两组聚类中心集合Cin和Cout。对于每一个亚细胞都可以生成一组这样的集合。
3) 标记间改进的K-means 算法中常数α 设定为0.15, 进一步优化Cin, 组成ML-RBF 的隐层中心,所有的Cout将用于结果调整。
4)通过隐层中心和属于亚细胞i的训练样本计算出ML-RBF 模型的权重。采用梯度下降算法调整权重和平滑指数,最后得到一个CL-RBF 分类器。
5)将测试样本特征向量作为输入送入分类器,分类器会给出一个或者多个预测结果,采用基于聚类优化的结果集调整策略以及调整预测结果。
6)若预测结果与实际的亚细胞位置相同,则预测正确,否则预测错误。
7) 预测完毕后将测试样本放回样本集合中并取出下一组子集作为测试样本,其余样本作为训练样本,再次训练出一个CL-RBF 分类器,并用测试样本进行测试。 以此类推直至所有子集测试完毕。
2 结果与分析
对比算法选取了ML-KNN 算法、LIFT 算法[20]、MLNB 算法[21]和ML-RBF 算法。 不同的分类预测算法在不同的数据集上单独运行50 次, 采用十折交叉实验,模型每运行完10 次,数据集会重新分成10个子集,评价指标取平均值。
我们使用多标记学习当中经常用到的5 个评价算法,汉明损失(Hamming loss)、单错误(one-error)、覆盖范围(coverage)、排位损失(ranking loss)和平均查准率(average precision)。 其中汉明损失、单错误、覆盖范围和排位损失这4 个评价指标越小,说明算法的效果越好。 而平均查准率,我们期望越大越好。由于5 种评价指标的考察方向不同,所以不能保证算法在所有的指标上都有很好的结果。
表5 为5 种多目标学习算法在革兰氏阳性菌464上的实验结果。 可以看出,ML-RBF 算法在革兰氏阳性菌464 数据集上的性能较好。 除了汉明损失这个指标,CL-RBF 算法在其他4 个指标上都最优秀,覆盖范围上比ML-RBF 算法降低了两个百分点,平均查准率达到了85.37%。
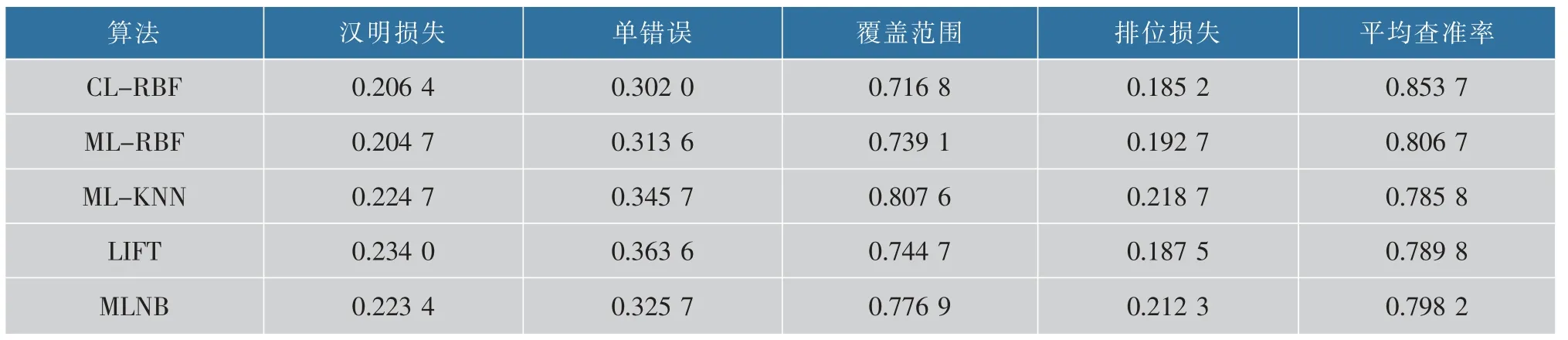
表5 不同多目标算法在革兰氏阳性菌464 上的性能比较Table 5 Performance of different multi-learning algorithms on gram-positive464 dataset
根据表6 的评价指标显示,在Gram-positive515数据集上,LIFT 和ML-RBF 的5 个评价指标也比较不错, 其中LIFT 算法出现单错误的概率最低,为24.41%。 CL-RBF 和ML-RBF 算法在其他4 个指标上都要优于其他的比对算法, 而LIFT 在这4 个指标上的表现紧随其后。 MLNB 在前4 种指标中都达到了最大值,并且在平均查准率上最小,因此MLNB在该数据集上的表现是最差的。
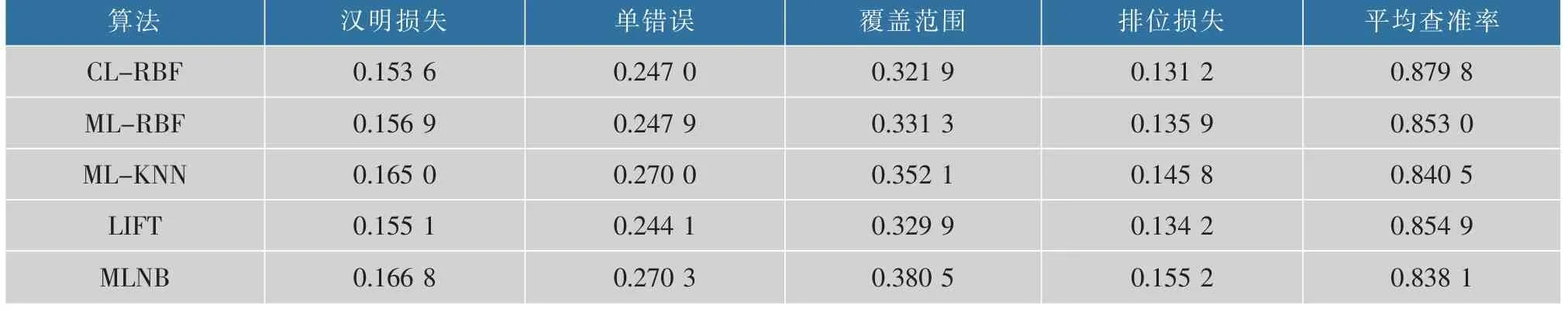
表6 不同多目标算法在革兰氏阳性菌515 上的性能比较Table 6 Performance of different multi-learning algorithms on gram-positive515 dataset
从表7 可知,ML-RBF 预测算法在数据集革兰氏阴性菌833 上的评价指标同样优于ML-KNN、LIFT和MLNB3 种算法, 而且经过改进后,CL-RBF 的指标数据都得到了提升,达到了最优值。 ML-KNN 算法在革兰氏阴性菌833 上的表现最差,在单错误、覆盖范围和排位损失都是最大的,而在平均查准率上是最小的。
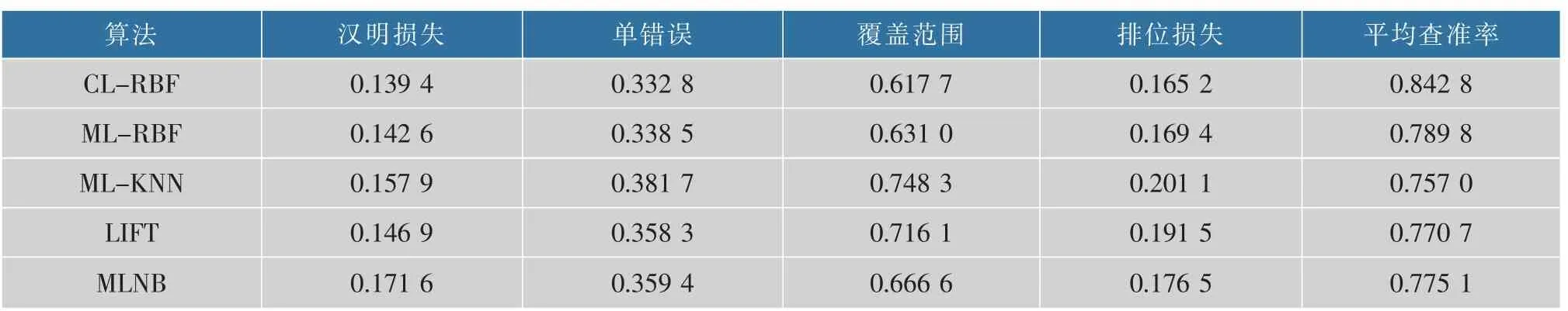
表7 不同多目标算法在革兰氏阴性菌833 上的性能比较Table 7 Performance of different multi-learning algorithms on gram-negative833 dataset
从表8 可知,ML-RBF 在该数据集上的表现仍旧很不错, 除了LIFT 在汉明损失这个指标上最优秀,CL-RBF 在这4 个评价指标上都达到了最优,ML-RBF 次之。ML-KNN 和MLND 算法在5 个评价指标上的表现都较差。
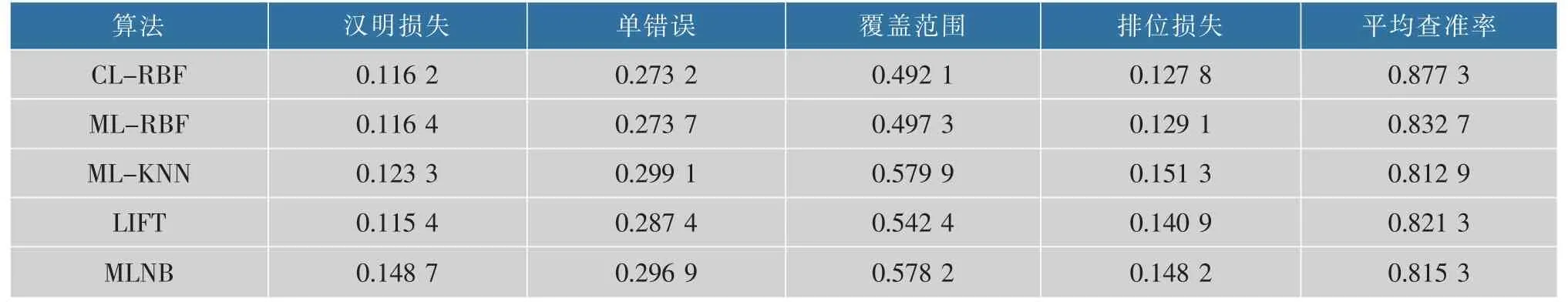
表8 不同多目标算法在革兰氏阴性菌1392 上的性能比较Table 8 Performance of different multi-learning algorithms on gram-negative1392 dataset
3 结 语
本研究在ML-RBF 算法的基础上,提出CL-RBF算法。 算法对隐层中心选取、参数计算和结果集处理分别进行了调整。 通过轮廓系数优化隐层中心的个数, 采用传统的K-means 聚类计算隐层中心的值,针对不同标记间隐层中心距离较近的情况进行了处理。 采用自适应的梯度下降算法对平滑指数和链接权重进行调整,充分考虑待测样本与该标记的隐层中心、不属于该标记的训练样本的聚类中心之间的关系,来进一步调整最终的预测值。 选取一个基于词袋模型和AAC 相结合的方法作为本研究的特征提取方法。 实验选取了LIFT、MLNB、ML-KNN和ML-RBF 4 种算法与CL-RBF 算法在4 个多标记数据集上进行对比实验。 从实验结果可以看出,在革兰氏阳性菌和革兰氏阴性菌的蛋白质亚细胞定位预测上,LIFT 算法、ML-RBF 算法和CL-RBF算法不论是在汉明损失、单错误、覆盖范围、排位损失还是平均准确率上,表现都要优于ML-KNN 算法和MLNB 算法。 三者的性能排序是LIFT 算法<MLRBF 算法<CL-RBF 算法,ML-RBF 经过改进后能在绝大部分评价指标上获得比较好的结果。 对比其他算法,改进的ML-RBF 算法在4 个多标记数据集上都取得了最优的覆盖范围、 排位损失和平均查准率。 这些结果证明CL-RBF 是一种较为有效的蛋白质亚细胞定位预测算法。
