苯并芘DNA加合物anti-BPDE-N2-dG四种立体异构体的合成及色谱分离
2020-05-08赵旭霞郭娅男郑速进张加玲阎小青
赵旭霞,冯 蕊,郭娅男,郑速进,张加玲,阎小青
(山西医科大学 公共卫生学院 卫生检验教研室,山西 太原 030001)
多环芳烃(PAHs)是一类广泛存在于环境的持久性有机污染物,主要来源于煤、石油等有机化合物的不完全燃烧,也会产生于烧烤食物以及沥青、焦油等的生产过程中。其中,苯并[a]芘(B[a]P)是最为典型的一种多环芳烃,具有极强的致畸、致癌和致突变性,在进入人体后经过一系列代谢活化作用生成邻二醇环氧苯并芘(BPDE)[1-3],此代谢产物有(±)anti-BPDE和(±)syn-BPDE两对对映异构体(anti-BPDE与syn-BPDE之间为非对映异构体)。大量的研究表明(±)anti-BPDE比其立体异构体(±)syn-BPDE更具诱变性[4-7],(±)anti-BPDE主要与生物体DNA中脱氧鸟苷的N-2位点结合[1-4],生成BPDE的脱氧鸟苷加合物(anti-BPDE-N2-dG)。此加合物是化学物质致突变、致癌进程中一个重要的启动因子[8],既可以作为暴露标志物,反映毒物在靶位的实际暴露剂量,又可以作为效应标志物,反映DNA受到有毒化学物质损伤的程度,对研究环境和职业B[a]P暴露的早发现、早预防有重要意义[9-10]。anti-BPDE-N2-dG含有两个手性碳原子,存在对映异构和顺反异构现象,主要有trans(-)、trans(+)、cis(+)、cis(-)-anti-BPDE-N2-dG四种立体异构(如图1所示)[11],其中,trans(-)与trans(+)-anti-BPDE-N2-dG,cis(+)与cis(-)-anti-BPDE-N2-dG为两对对映异构体(手性异构体),而trans与cis-anti-BPDE-N2-dG为顺反异构体。研究表明这四种立体异构体的生物学活性存在显著差异[12]。因此,对anti-BPDE-N2-dG加合物的四种立体异构体进行准确地定性和定量分析,对B[a]P暴露监测及其致癌、致突变的作用机理研究非常重要。
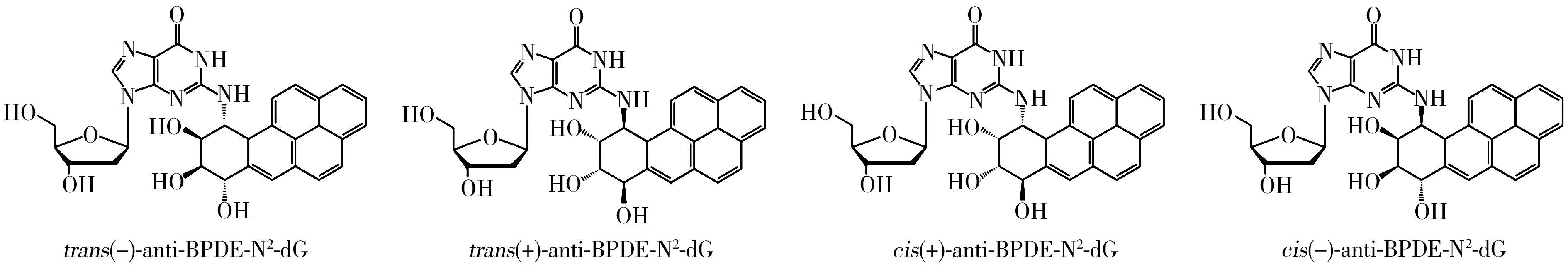
图1 anti-BPDE-N2-dG立体异构体的分子结构Fig.1 Molecular structures of anti-BPDE-N2-dG stereoisomers
环境中多环芳烃的暴露水平极低,所以暴露后的生物体内相应形成的DNA加合物含量更低。大约108~1010个核苷酸中含有1个加合物[4],如何从大量的正常核苷中检出痕量的特定加合物是当今分析技术面临的挑战。目前,关于苯并芘DNA加合物的检测发展了多种方法[13-14],包括32P标记法、免疫吸附法、核磁共振法、荧光检测法,但这些方法均无法对加合物的立体异构体进行表征。相比之下,高效液相色谱-质谱联用法(HPLC-MS/MS)不仅拥有可与32P后标记法相媲美的灵敏度,而且还可以提供加合物的结构信息,因此受到了更多关注[15-19],但在使用该方法定量检测BPDE-DNA加合物时,需合成相应的标准品。目前,文献报道了BPDE-DNA加合物的不同合成方法[15-18],其中,王海林课题组[15]创新性地通过anti-BPDE与2′-脱氧鸟苷(Deoxyguanosine,dG)直接反应后,用固相萃取(SPE)和高效液相色谱(HPLC)对反应产物进行提纯制得anti-BPDE-N2-dG四种立体异构体。该方法与以往的合成方法相比,具有无需酶解、反应时间短、BPDE用量少等优点。但该合成方法利用常规苯基柱需要85~100 min[15]才可将anti-BPDE-N2-dG四种立体异构体实现色谱分离提纯。另外,在利用HPLC-MS/MS对anti-BPDE-N2-dG四种立体异构体进行检测时,虽然质谱的选择性监测可排除副产物四醇-B[a]P的干扰,缩短色谱分离时间,但仍需45~60 min才能将四种立体异构体分离并检测[18-19],且流速快,分流检测使得样品用量大,分离度较小。
五氟苯基柱(PFP)是在硅胶基质上键合的五氟苯基硅烷键合相,由于五氟苯基的存在,化合物可与键合相之间产生π-π共轭、偶极-偶极、氢键等作用[20],因此PFP柱的选择性有别于C18柱和苯基柱。本研究首次将PFP应用于anti-BPDE-N2-dG四种立体异构体的分离,结果表明,相对于常规C18色谱柱,anti-BPDE-N2-dG 四种立体异构体在 PFP色谱柱上的分离效果更好。通过对色谱条件的优化,可实现45 min内将四种立体异构体分离提纯。在使用HPLC-MS/MS检测时,30 min内即可完成检测,极大提高了分离与检测效率。
1 实验部分
1.1 仪器与试剂
高效液相色谱配二极管阵列(DAD)检测器(Thermo scientific U3000,美国Thermo公司);高效液相色谱-串联质谱(LCMS-8050,日本岛津公司);MOS 450圆二色光谱仪(法国BioLogic公司);TU-1901双光束紫外分光光度计(北京普析通用仪器有限公司);SCIENTZ-12N冷冻干燥机(宁波新芝生物科技股份有限公司);GI36DS高压灭菌锅(厦门致微仪器有限公司);Mikro 220R高速冷冻离心机(德国Hettich公司);PB-10 pH计(德国Sartorius公司);ME104E电子天平(瑞士Mettler-Toledo公司);RCT basic加热磁力搅拌器(德国IKA集团)。
(±)anti-BPDE(纯度>99%,美国MRIGlobal Chemical Carcinogen Repository);2′脱氧鸟苷(纯度98%,百灵威科技有限公司);小牛胸腺DNA(美国Sigma公司);脱氧核糖核酸酶Deoxyribonuclease I from bovine pancreas(美国Sigma公司);蛇毒磷酸二酯酶Phosphodiesterase I from Crotalus adamanteus venom(美国Sigma公司);Tris-base(上海Sangon Biotech);四氢呋喃、三乙胺、氯化钠、氯化钙、乙醚、无水乙醇、乙酸乙酯、浓盐酸(分析纯,天津市光复精细化工研究所);六水氯化镁(上海BBI Life Sciences);甲醇、乙腈(色谱纯,美国Thermo Fisher公司);甲酸(色谱纯,天津市致远化学试剂有限公司);超纯水(屈臣氏饮用水)。
1.2 anti-BPDE-N2-dG四种立体异构体标准品的制备
1.2.1 合成反应及固相萃取的初步纯化(±)anti-BPDE与小牛胸腺DNA反应生成BPDE-脱氧鸟苷酸化合物(BPDE adduct of deoxyguanosine monophosphate,BPDE-dGMP):参照Vouros[21]课题组的方法合成BPDE-dGMP。称取 0.9 mg(±)anti-BPDE溶于1 mL四氢呋喃;称取3.6 mg小牛胸腺DNA溶于5 mL 0.05 mol/L Tris-HCl(pH 6.8);将两者混匀,50 ℃避光孵育8 h。孵育结束,加乙酸乙酯-乙醚(5∶2,体积比)7 mL液液萃取,收集下层液体,重复3次。加入NaCl使其在溶液中的浓度为0.14 mol/L,摇匀,再加入2.5倍体积的无水乙醇,充分混匀,-22 ℃静置20~30 min后,-10 ℃ 12 000 r/min离心20 min,移走上清液,室温下干燥,复溶于0.05 mol/L含5 mmol/L Ca2+、4 mmol/L Mg2+的Tris-HCl(pH 8.6)缓冲溶液。加入DNA酶 800 U,蛇毒磷酸二酯酶(Snake 酶)0.1 U,37 ℃避光孵育20 h。随后将消化后的反应液进行固相萃取净化富集。
固相萃取:称取100 mg C18固相萃取吸附剂,甲醇活化并装入固相萃取小柱,5 mL水替换甲醇,取1 mL反应液通过固相萃取小柱,4 mL水溶液淋洗,抽干小柱(不滴水即可),4 mL 40%甲醇水缓慢洗脱小柱(靠重力流下),收集洗脱液并浓缩:N2吹干,复溶于500 μL甲醇,经0.45 μm PTFE针头滤器过滤后待测。
(±)anti-BPDE与dG反应合成BPDE-脱氧鸟苷加合物(±)anti-BPDE-N2-dG:参照王海林课题组的方法[15],称取0.1 mg (±)anti-BPDE溶于100 μL四氢呋喃-三乙胺(19∶1,体积比)溶液中;称取4.8 mg的2′脱氧鸟苷(dG)溶于1.2 mL 50 mmol/L Tris-HCl(pH 7.5)缓冲溶液中,将两者混匀,避光,室温反应48 h。
固相萃取:称取100 mg C18固相萃取吸附剂,甲醇活化并装入固相萃取小柱,5 mL水替换甲醇,将反应液通过固相萃取小柱,再分别用2 mL的超纯水、1 mL 10%甲醇水溶液、1 mL 30%甲醇水溶液淋洗,抽干小柱(不滴水即可)后,用2 mL甲醇洗脱小柱(靠重力流下),收集洗脱液并浓缩(室温挥发)至1 mL后待测。
1.2.2 高效液相色谱纯化合成的产物经固相萃取后不能去除副产物四醇-BaP的干扰,因此,需继续浓缩后经高效液相色谱仪(DAD检测器)进一步纯化。选用PFP色谱柱(250 mm×4.6 mm,5 μm),流动相为乙腈-0.1%甲酸水(22.5∶77.5,体积比),流速1.2 mL/min,柱温30 ℃,进样量20 μL,紫外检测波长345 nm。反复多次进样,并分别收集保留时间为22.887、28.090、30.430 、33.763 min的四种异构体流出物,经冷冻干燥后,分别复溶于500 μL的33%甲醇水溶液中,由圆二色谱仪对四种组分进行定性分析,确定四种立体异构体出峰顺序。将定性后的四种立体异构体分别用紫外光谱定量,测定345 nm处紫外吸光度,根据ε345 nm=3.43×104L/(mol·cm)算出各物质浓度[18]。
1.3 HPLC-MS/MS检测anti-BPDE-N2-dG四种立体异构体
色谱条件:PFPP色谱柱(250 mm×4.6 mm,5 μm);流动相:乙腈-0.1%甲酸水(28∶72),流速:0.5 mL/min;柱温:30 ℃;进样量:5 μL。
质谱条件:以正离子多反应监测(MRM)模式检测,碰撞电压:12 eV,母离子质荷比(m/z)为570.15,子离子m/z为454.10;采用电喷雾离子源(ESI+);雾化气:3 L/min,离子源加热气:10 L/min,干燥气:10 L/min,离子源温度:250 ℃,脱溶剂管温度:150 ℃;加热块温度:350 ℃;碰撞诱导解离气:270 kPa;采集时间(Dwell time):100 ms。
2 结果与讨论
2.1 anti-BPDE-N2-dG四种立体异构体的制备
2.1.1 anti-BPDE-N2-dG合成方法的选择使用HPLC-MS/MS定量检测苯并芘DNA加合物时,由于无市售的加合物标准品,需要合成相应的纯品。根据文献报道,苯并芘的脱氧鸟苷加合物主要有两种合成方法,一种是BPDE与小牛胸腺DNA反应生成BPDE的脱氧鸟苷酸加合物(BPDE-dGMP)[21],另一种为(±)anti-BPDE直接与2′脱氧鸟苷(dG)反应生成anti-BPDE-N2-dG[15]。本实验对两种方法进行了对比。第一种(具体步骤见“1.2.1”)中,BPDE与小牛胸腺DNA反应后,酶解至单核苷酸,经固相萃取纯化,质谱子离子扫描[21],验证合成BPDE-dGMP。但此合成过程中(±)anti-BPDE的用量大,因为DNA中除鸟嘌呤外,还有其他碱基会与BPDE反应从而消耗BPDE;合成过程需酶解,耗时且酶价格较昂贵,而繁琐的反应过程也使得实验的重复性较差。第二种方法选择(±)anti-BPDE与dG直接反应生成(±)anti-BPDE-N2-dG,经固相萃取初步纯化后,采用质谱子离子扫描[15,17]验证合成的anti-BPDE-N2-dG。该方法中(±)anti-BPDE的用量少,从经济和安全性角度均非常有益,且反应无需酶解,过程简单方便,因此,本实验选用该方法合成BPDE的DNA加合物标准品。
2.1.2 高效液相色谱纯化目前,文献报道的纯化方法采用苯基柱需85 min[15]才能将anti-BPDE-N2-dG两对对映体实现色谱分离提纯,时间较长。本实验采用常规的C18色谱柱(250 mm×4.6 mm, 5 μm)对其进行分离,结果显示在流动相条件为乙腈-水(18.5∶81.5,体积比),流速0.75 mL/min,需要160 min才可实现六种物质(BPDE-N2-dG四种立体异构体和两种四醇-BaP)的分离,时间很长。因此,本实验尝试将不同于C18柱和苯基柱作用机制的五氟苯基柱(PFP柱)应用于两对对映体的分离,并与常规C18色谱柱进行对比,结果如图2所示。图中星形标记的为anti-BPDE-N2-dG立体异构体,峰1和峰2为副产物四醇-BaP的异构体。结果表明:在相同流动相条件下(乙腈-水,体积比为26∶74,流速1.2 mL/min),345 nm处紫外光谱检测,anti-BPDE-N2-dG的四种立体异构体在C18柱上只能看到两个峰(图2A),而在相同规格的PFP色谱柱上(图2B)可观察到四种立体异构体的分离。
与中性或碱性流动相相比,酸性流动相可增强cis-(+)-anti-BPDE-N2-dG的稳定性,并提高四种立体异构体的分离度[19]。因此,在水中加入0.1%甲酸作为流动相,PFP色谱柱在相同流动相比例下(乙腈-0.1%甲酸水,体积比26∶74,流速1.2 mL/min),可观察到anti-BPDE-N2-dG四种立体异构体中的第2、3、4峰的分离度明显增大,且第3与第4个峰已达到基线分离(图2D)。在此酸性流动相条件下,anti-BPDE-N2-dG的四种立体异构体在C18柱上可分离出三个峰(图2C),较中性流动相的两个峰(图2A)有明显改善,但分离效果不及PFP色谱柱(图2D)。
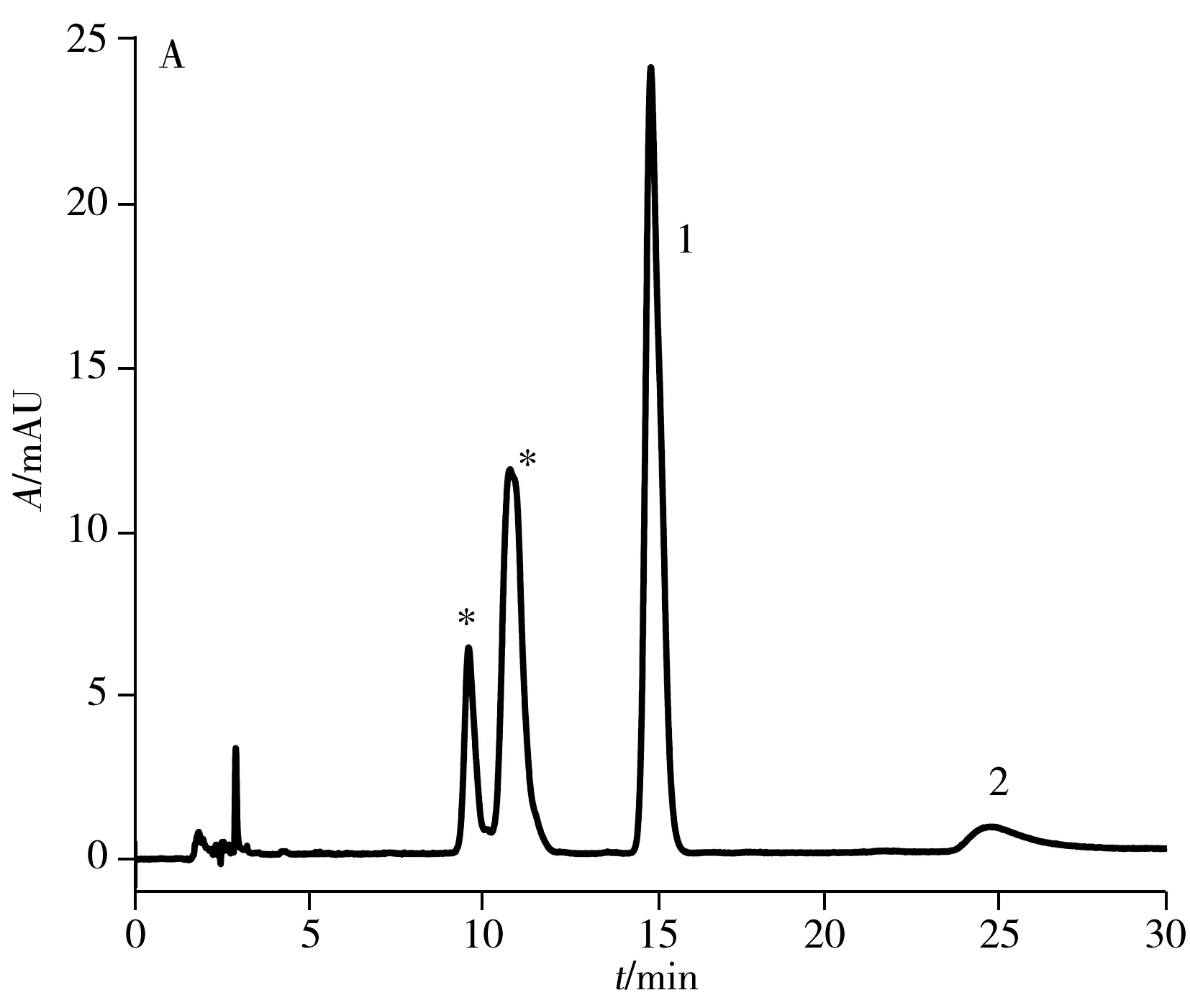
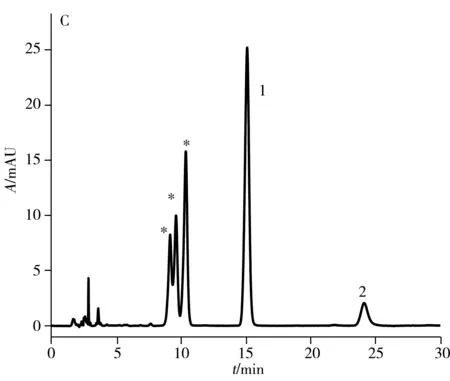
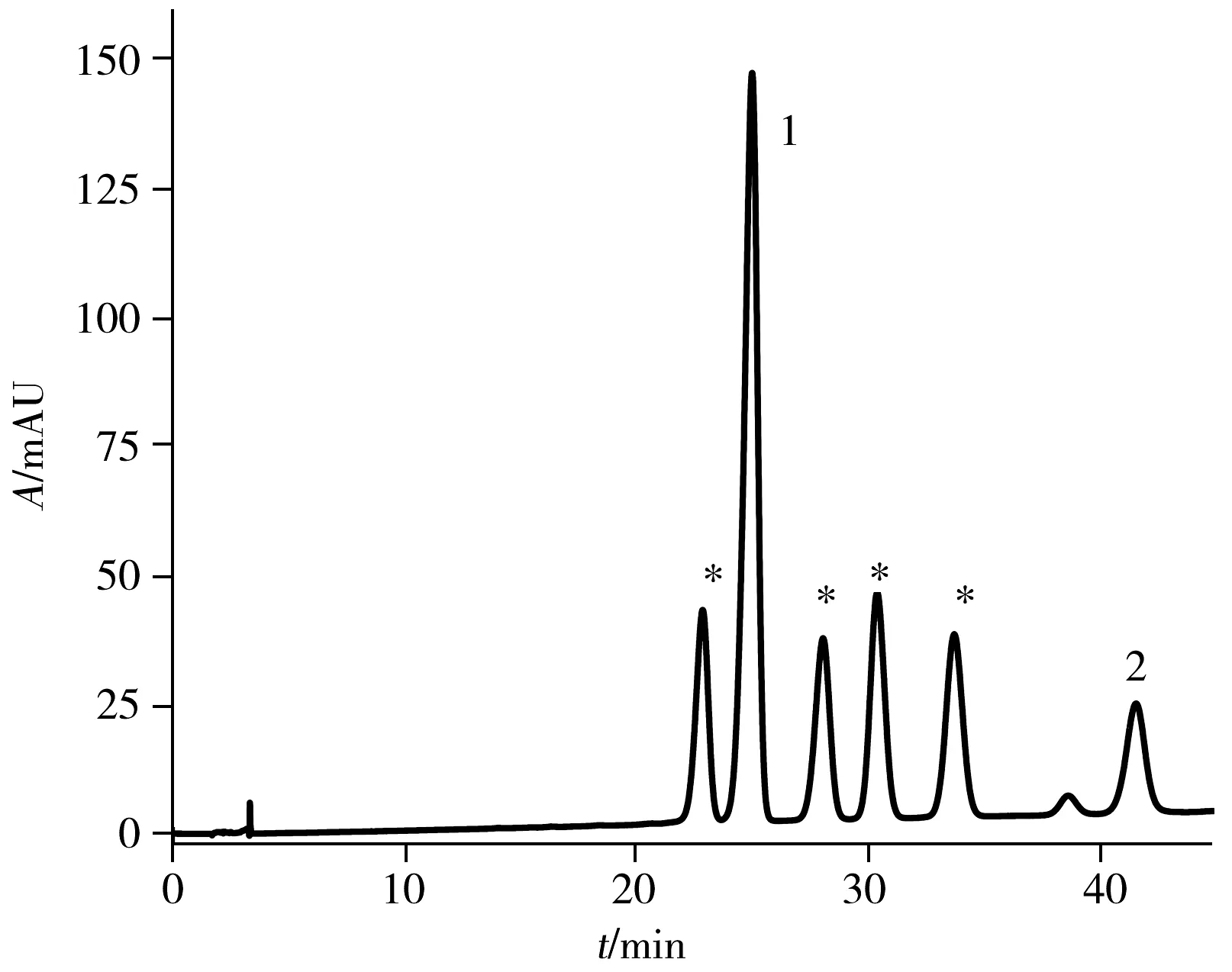
图3 PFP色谱柱上分离anti-BPDE-N2-dG四种立体 异构体及两种四醇-B[a]P的液相色谱图Fig.3 HPLC chromatogram of four anti-BPDE-N2-dG stereoisomers and two BPDE tetrols obtained on PFP column the peaks of 1 and 2 are two BPDE tetrols,and the peaks labeled with asterisk are anti-BPDE-N2-dG stereoisomers;mobile phase: acetonitrile-0.1% formic acid(22.5∶77.5);λ=345 nm
为了进一步增加四种立体异构体在PFP色谱柱上的分离度,继续调整流动相的比例,发现随着有机相比例的减少,四种立体异构体的分离度增加,六种物质(anti-BPDE-N2-dG顺反两对异构体和两种四醇-B[a]P)的保留时间均向后移,但anti-BPDE-N2-dG后移速度快于四醇-B[a]P。因此,anti-BPDE-N2-dG与四醇-B[a]P的峰出现重叠,流动相比例为乙腈-0.1%甲酸水(22.5∶77.5),流速1.2 mL/min时,45 min内可将六种物质完全分离开,分离度均大于2(如图3)。分别收集anti-BPDE-N2-dG四种立体异构体,即保留时间分别为22.887、28.090、30.430、33.763 min的流出物并冷冻干燥去除溶剂,复溶于33%甲醇水溶液中进行圆二色谱(CD)及紫外吸收光谱分析。
2.1.3 anti-BPDE-N2-dG合成条件的优化为了提高anti-BPDE-N2-dG的合成产量,本实验对(±)anti-BPDE直接与dG反应的温度和时间进行了考察。控制其他反应条件不变,将相同的两份样品分别置于室温(23.6 ℃)与37 ℃,避光反应24 h;固相萃取初步纯化后,HPLC-DAD检测(乙腈-水,26∶74,C18柱)anti-BPDE-N2-dG与四醇-B[a]P的含量(如图2A),并比较其峰高。结果如表1所示:表中anti-BPDE-N2-dG为图2A中保留时间为11 min的峰,四醇-B[a]P 1为图中保留时间为15 min的峰1,四醇-B[a]P 2为图中保留时间为25 min的峰2。经两独立样本t检验显示,室温下anti-BPDE-N2-dG的产量较37 ℃高(P<0.05)(表1),而四醇-B[a]P的量下降,表明室温下更有利于主反应的进行。控制其他反应条件不变,仅将样品在室温反应时间由24 h延长至48 h,结果显示:(±)anti-BPDE与dG反应48 h时,anti-BPDE-N2-dG与四醇-BaP的峰高较24 h均提高2倍多(见表1)。为了提高anti-BPDE-N2-dG的合成产量,本实验将反应条件定为室温反应48 h。
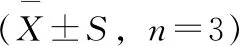

Experimental conditionAanti-BPDE-N2-dG(mAU)ABPDE tetrol 1(mAU)ABPDE tetrol 2(mAU)37 ℃,24 h52.17±0.28 74.17±0.73 2.56±0.37 23.6 ℃,24 h56.42±0.68 71.02±0.48 1.37±0.35 23.6 ℃,48 h 142.2±3.01 195.0±0.99 3.35±0.18
2.2 anti-BPDE-N2-dG立体异构体的表征及定量
在HPLC-DAD上,anti-BPDE-N2-dG与副产物四醇-B[a]P的紫外吸收光谱很相似,但仍可通过比较紫外图谱中280~282 nm和344~347 nm处相对吸收强度来区分anti-BPDE-N2-dG与四醇-B[a]P[15]。如图4所示,anti-BPDE-N2-dG的紫外吸收在280~282 nm处相对较高,而两种四醇-B[a]P的紫外吸收在344 nm处相对较高。此外,根据最长波长的紫外吸收峰位置还可粗略区分顺反异构体,cis(+)和cis(-)对映异构体最长的紫外吸收峰位于347 nm,而trans(+)和trans(-)对映异构体最长的紫外吸收峰位于346 nm处,cis-anti-BPDE-N2-dG相比于trans-anti-BPDE-N2-dG略有红移[15,19]。
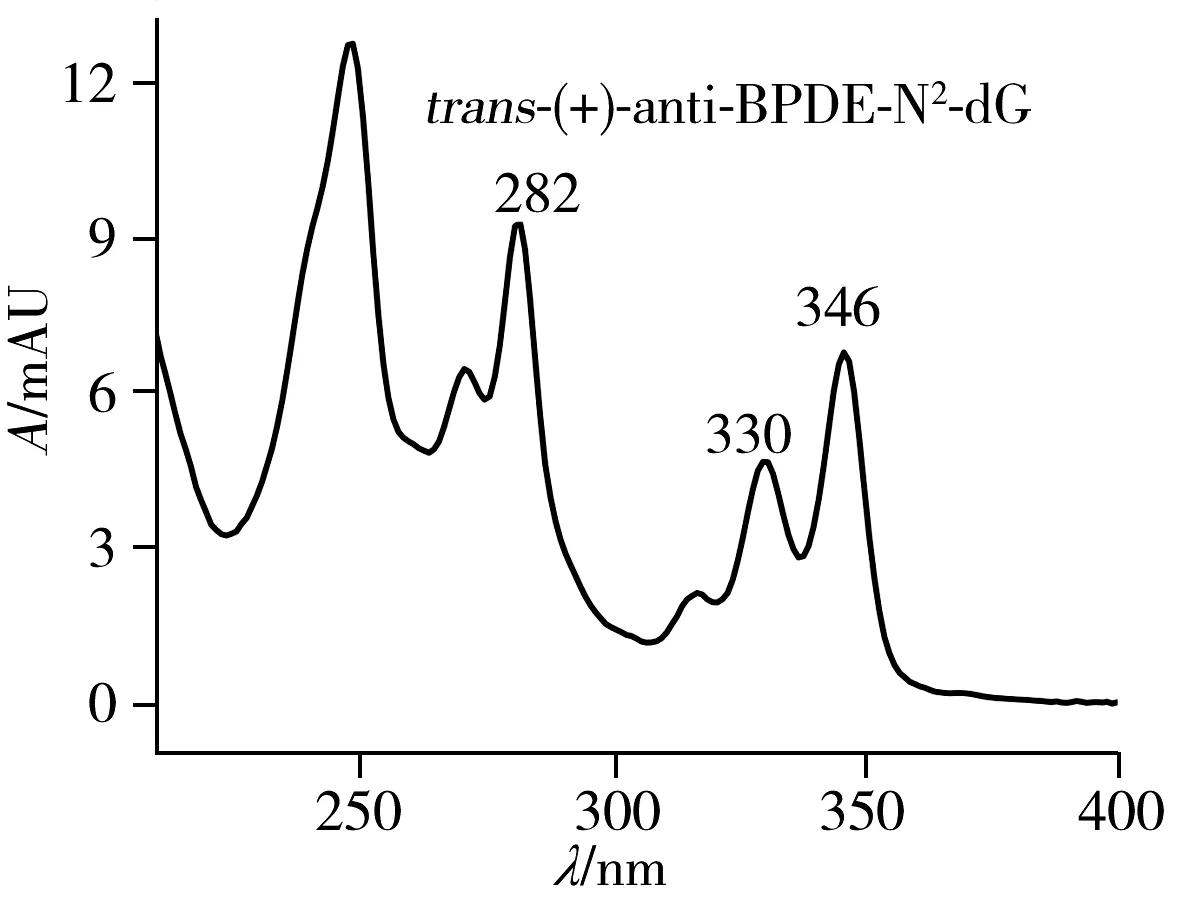
为了准确区分四种立体异构体,以及其中的对映异构体,将收集的四种组分复溶于33%甲醇水溶液中进行圆二色谱(CD)表征。对映异构体加合物trans-(+)和trans-(-),cis-(+)和cis-(-)的CD光谱相似,但符号相反。trans-(+)-和cis-(-)-anti-BPDE-N2-dG均具有10S的绝对构型,且对其最强的CD波段(250 nm)均表现出正信号。而trans-(-)-和cis-(+)-anti-BPDE-N2-dG均具有10R的绝对构型,且在相同的CD波段(250 nm)表现出负信号(图5)。研究结果与文献一致[19,22]。因此,确定PFP色谱柱分离anti-BPDE-N2-dG四种立体异构体的出峰顺序为trans(-)(22.887 min)、trans(+)(28.090 min)、cis(+)(30.430 min)、cis(-)(33.763 min)-anti-BPDE-N2-dG(图3)。将复溶于33%甲醇水溶液的各异构体溶液,测定其在345 nm处的紫外吸光度,可根据ε345 nm=3.43×104L/(mol·cm)算出各物质的浓度[18]。
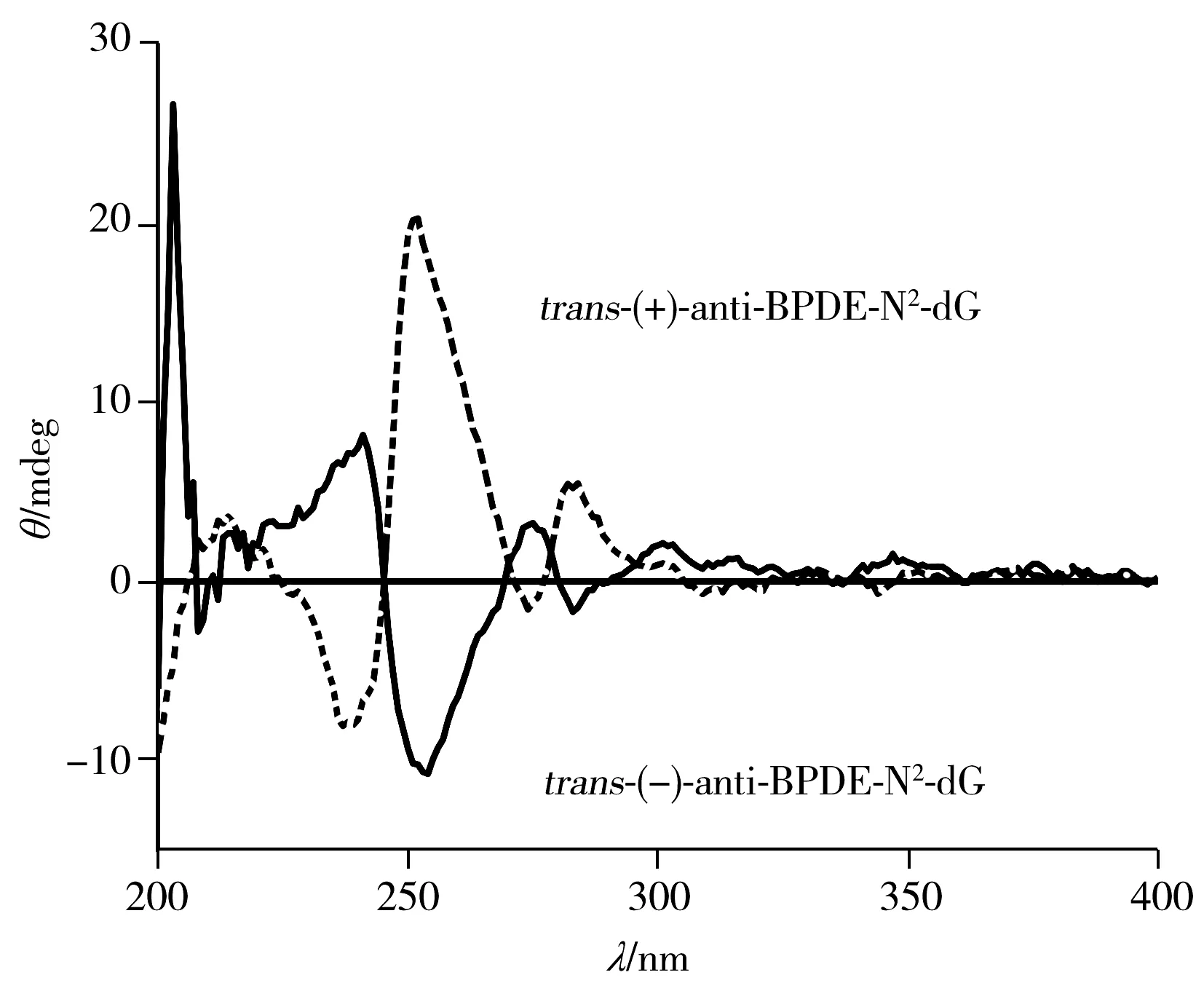

图6 HPLC-MS/MS检测anti-BPDE-N2-dG 四种立体异构体的色谱图Fig.6 Chromatogram for separation of four anti-BPDE-N2-dG stereoisomers recorded by HPLC-MS/MS
2.3 HPLC-MS/MS色谱分离条件的选择
利用HPLC-MS/MS对样品进行检测时,质谱在离子选择模式下检测可排除四醇-B[a]P的干扰,因此可不考虑四醇-B[a]P对色谱分离的影响,将anti-BPDE-N2-dG四种立体异构体分开即可满足检测条件。如果将HPLC-DAD分离anti-BPDE-N2-dG的条件:乙腈-0.1%甲酸水(22.5∶77.5),1.2 mL/min,PFP柱(250 mm×4.6 mm,5 μm)直接应用于HPLC-MS/MS,1.2 mL/min流速的对于质谱过高,不利于雾化,灵敏度偏低,故将流速设为0.5mL/min,调整流动相比例为乙腈-0.1%甲酸水(28∶72),柱温30 ℃,即可在30 min内分离四种立体异构体(图6),最小分离度可达0.995。与相同规格的苯基柱需要60 min才能将anti-BPDE-N2-dG四种立体异构体分离相比,大大缩短了分离时间,提高了分离效率,节约了分离成本。
3 结 论
本文对现有的合成纯化anti-BPDE-N2-dG四种立体异构体(包含两对对映异构体)方法进行了改进。通过优化反应温度和时间,提高了反应产量,为定量检测生物体苯并芘DNA加合物提供了标准品;并首次将五氟苯基色谱柱应用于anti-BPDE-N2-dG四种立体异构体的色谱分离,利用HPLC可以实现45 min内分离提纯四种立体异构体,建立了一种快速纯化与分离anti-BPDE-N2-dG四种立体异构体的方法。利用HPLC-MS/MS检测四种anti-BPDE-N2-dG立体异构体标准品时,使用常规的五氟苯基色谱柱可在30 min内完成分离,大大提高了检测效率。制备纯化以及检测分离anti-BPDE-N2-dG均可在同一色谱柱上完成,节约了分离成本。
