PAX3基因突变相关Waardenburg综合征家系及散发患者基因型与表型特征分析
2020-04-27李进关静张静谢林怡熊芬兰兰王秋菊
李进关静张静谢林怡熊芬兰兰王秋菊*
1浙江中医药大学(杭州310053)
2中国人民解放军总医院耳鼻咽喉头颈外科医学部,国家耳鼻咽喉疾病临床医学研究中心,解放军耳鼻咽喉研究所,聋病教育部重点实验室;聋病防治北京市重点实验室(北京100853)
Waardenburg Syndrome(WS),即瓦登伯格综合征,又称色素-听力综合征,是常见的耳聋综合征之一,发病率约1/42000,占先天性聋的2%-5%[1]。主要临床症状包括先天性感音神经性聋、虹膜色素异常、毛发皮肤色素异常等;根据不同特征性表现分为Ⅰ-Ⅳ四个亚型,分别为WS1-WS4,WS1伴內眦异位,WS2不伴內眦异位,WS3合并上肢异常,又称Klein-Waardenburg综合征,WS4常伴先天性巨结肠等胃肠道畸形及周围神经脱髓鞘病变、中枢髓鞘形成障碍等神经病学改变,又称Waardenburg-Shah综合征(https://ghr.nlm.nih.gov/condition/waardenburg-syndrome#)。WS遗传方式包括常染色体显性遗传和常染色体隐性遗传,目前已发现6个与WS相关的致病基因,分别为MITF、PAX3、EDNRB、EDN3、SOX10及SNAI2(https://hereditaryhearingloss.org/ws),且不同基因与不同亚型相关,其中PAX3基因(paired box 3)(OMIM:606597)是发现的第一个WS相关基因,还与颅面-聋-手综合征(craniofacial-deafness-hand syndrome,CDHS)及腺泡状横纹肌肉瘤相关(https://www.ncbi.nlm.nih.gov/gene/5077)。本研究在12例疑似WS患者中检出5例PAX3基因的变异,本文将对这5个家庭的基因型与表型特征进行如下分析。
1 研究对象
对本课题组2017-2018年间收集的疑似WS患者进行耳聋基因检测,对检出PAX3基因突变的5名患者及其家庭进行分析(图1-4)。所收集的5名患者分别来自辽宁、山东、江苏、河北,4名具有家族遗传史,1名为散发患者;5名患者包括1名男性及4名女性;就诊年龄4名为婴幼儿或儿童(0-6岁),1名为成年人(31岁)。4个有家族遗传史的家系中,共收集先证者及家庭成员血样18份,其中有WS相关表现者10人;1个疑似新生突变患者家庭中,仅收集到先证者及其母亲血样。所有研究对象均为汉族,均签署知情同意书。

图1 家系1基因型与表型A.家系图:箭头所指为先证者;WT表示基因检测无突变,Het表示杂合突变;B.眼睛外观图:从上至下依次为Ⅰ-1、Ⅱ-2、Ⅲ-1眼睛外观图,分别为双侧蓝虹膜、左侧蓝虹膜、右侧蓝虹膜,Ⅱ-2、Ⅲ-1双眼间距较宽;C.Ⅰ-1、Ⅱ-1的Sanger测序验证结果;D.II-2纯音测听,提示左侧极重度听力损失Fig.1 Genotype and phenotype of Family 1.A.Pedigree:The proband is indicated by an arrow.WT and Het represents wild type and Heterozygous,respectively.B.Appearance of patients:Ⅰ-1 shows bilateral blue iris,Ⅱ-2 shows left blue iris andⅢ-1 shows right blue iris.Ⅱ-2 andⅢ-1 show widened interpupillary distance.C.Sequencing chromatograms ofⅠ-1,Ⅱ-1.D.Audiogram of II-2 shows severe hearing loss in left ear.

图2 家系2基因型与表型A.家系图:箭头所指为先证者;WT表示基因检测无突变,Het表示杂合突变;B.Sanger测序结果,从上至下依次为Ⅰ-2、Ⅱ-1;C.表示先证者纯音测听,提示双侧极重度感音神经性听力损失Fig.2 Genotype and phenotype of Family 2.A.Pedigree:The proband is indicated by an arrow.WT and Het represents wild type and Heterozygous,respectively.B.Sequencing chromatograms ofⅠ-2 andⅡ-1 shows WT and Het genotype,respectively.C.Audiogram of II-1 show bilateral severe hearing loss.
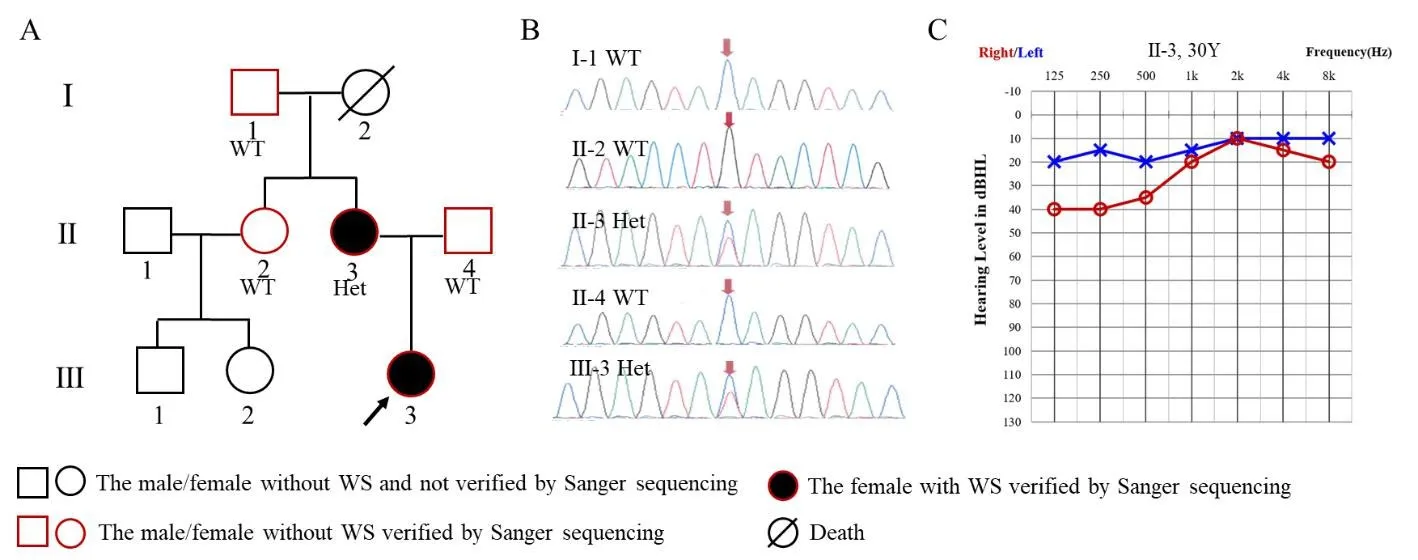
图3 家系3基因型与表型A.家系图:箭头所指为先证者;WT表示基因检测无突变,Het表示杂合突变;B.Sanger测序结果,从上至下依次为Ⅰ-1、Ⅱ-2、Ⅱ-3、Ⅱ-4、Ⅲ-3;C.Ⅱ-3纯音测听结果显示右侧低频听力下降Fig.3 Genotype and phenotype of Family 3.A.Pedigree:The proband is indicated by an arrow.WT and Het represents wild type and Heterozygous,respectively.B.Sequencing chromatograms ofⅠ-1,Ⅱ-2,Ⅱ-3,Ⅱ-4 and Ⅲ-3.C.Audiogram of II-3 shows hearing loss at low frequency in right ear.

图4 家系4-5基因型与表型:箭头所指为先证者;WT表示基因检测无突变,Het表示杂合突变;A.家系4家系图,III-2为先证者,先证者、II-2及II-3有WS表现;B.家系4中II-3纯音测听提示左侧极重度感音神经性听力损失;C.家系4样本Sanger测序结果D.家系5家系图,II-1为先证者,先证者及I-1有WS表现;E.家系5样本Sanger测序结果;F.保守性预测,可见该位置氨基酸高度保守Fig.4 Genotype and phenotype of Family 4 and Family 5.The proband is indicated by an arrow.WT and Het represents wild type and Heterozygous,respectively.A.Pedigree of the Family 4.The proband(III-2),II-2 and II-3 are WS patients.B.Audiogram of II-3 from Family 4 shows severe hearing loss in left ear.C.Sequencing chromatograms ofⅡ-2,Ⅱ-3,Ⅱ-4 andⅢ-2 from Family 4.D.Pedigree of the Family 5.The proband(II-1)and I-1 are WS patients.E.Sequencing chromatograms of I-1,II-1 andⅡ-1 from Family 5.F.The arginine,terminated at position of 222 of PAX3,is evolutionarily conserved among different species.
2 研究方法
2.1 临床资料收集及听力学评估
对患者进行详细问诊及体格检查,记录病史及家族史,包括皮肤、毛发、四肢、眼科、耳科等,并绘制家系图谱;使用抗凝管抽取4-5ml静脉血,置于-80℃保存备用;对患者进行听力学检查,包括纯音测听、声导抗、畸变产物耳声发射(DPOAE)、听性脑干反应(ABR)潜伏期,对无法配合主观测听的儿童及婴幼儿增加听性脑干反应阈值、40Hz听觉事件相关电位(40Hz AERP)及听觉稳态反应(ASSR)。根据500、1000、2000、4000Hz平均听阈计算听力损失程度,分为轻度(26-40 dB HL)、中度(41-55 dB HL)、中重度(56-70 dB HL)、重度(71-90 dB HL)以及极重度(>90 dB HL)听力损失。
2.2 基因检测及Sanger验证
抽取先证者及家系成员(共20名)外周静脉血,利用天根生化科技(北京)有限公司的血液基因组DNA提取试剂盒进行DNA提取,对先证者进行已知的127个候选基因的遗传性耳聋基因检测(NGS Panel),并在先证者及家系相关成员中进行Sanger验证。依据《遗传变异分类标准与指南》(ACMG指南)[2]明确突变的致病性。进行基因型表型的对比分析。
3 结果
3.1 临床表型
5名PAX3突变相关WS中,4名为家族遗传性,1名否认家族遗传史,疑似新发变异。4个家系中,先证者均为极重度感音神经性听力损失,2名双侧,2名单侧;均存在虹膜异色,2名双侧,2名单侧;3名存在内眦间距异常。对先证者及亲属进行追访,共记录10名患者,8名(80%,8/10)存在听力损失,其中6名(75%,6/8)为单侧;9名(90%,9/10)存在虹膜异色,其中4名(44.4%,4/9)为单侧。另外,部分患者存在额白发、一字眉等表现,均不存在皮肤色素异常。疑似新发变异患者为双侧极重度感音神经性听力损失,双侧虹膜色素异常,且有鼻部形态异常及手指弯曲、肘关节突出(表1)。
3.2 基因检测结果及分析
5例PAX3相关WS患者中,共检测到4种突变,包括1例内含子剪切位点变异,1例错义突变及3例无义突变;5例患者中,4例有明确家族史,1例无家族史。对这4个具有家族遗传性的先证者及其家属共18名(10例患者,8例正常人)进行Sanger验证,结果表明有WS表现者均为杂合突变,基因型与表型共分离,符合显性遗传特征。家系1(图1)为PAX3基因6号内含子上发生的碱基替换c.958+75A>G(NM_181457.3),该变异尚无文献报道,根据ACMG指南,判断为意义未明变异。家系3检出c.790C>T(p.Gln264*,NM_181457.3),家系4和5均检出c.664C>T(p.Arg222*,NM_001127366),这2种无义突变既往多次在WS1患者中报道[3-5],为致病变异。家系2(图2)先证者为PAX3基因c.141C>A(p.Asn47Lys,NM_181457.3)错义突变,该变异曾在一个CDHS家系中报道[6],而相同位置p.Asn47His曾在WS3[7]患者中检测到。值得注意的是,此患者否认家族史,我们在已获得的母亲样本中未检测到相同变异,因患者父亲样本无法提供,我们无法确认突变来源。但并不排除患者的致病突变为新生突变的可能性。
4 讨论
PAX3基因位于2号染色体,编码PAX3转录因子,具有高度保守性[8]。PAX3转录因子包括N端DNA结合区(含配对盒结构域PD和同源结构域HD)、八肽基序及C端转录激活区,DNA结合区行使主要功能[9]。PAX3基因突变可造成黑素细胞合成异常、神经嵴发育异常,从而造成毛发皮肤低色素、虹膜异色以及颅面骨骼、牙齿及四肢等异常,故可表现为WS或CDHS。约80%的WS1患者可检出PAX3的突变,WS3也与PAX3相关[8,10];目前已报道的PAX3的WS相关变异有100余种(https://grenada.lumc.nl/LOVD2/WS/home.php?select_dp=PAX3),包括错义突变、无义突变、剪切位点变异、移码突变等,突变多位于第2~6外显子[11],可能改变了PD和HD功能的完整性,从而影响PAX3转录因子的功能,且纯合突变的表型多比杂合突变严重[12]。本文通过对12例疑似WS患者进行基因检测,发现5例PAX3基因突变相关患者,显性遗传4例及疑似新生变异1例,包括4种突变类型,分别为c.958+75A>G、c.141C>A(p.Asn47Lys)、c.790C>T(p.Gln264*)及c.664C>T(p.Arg222*),其中c.958+75A>G为本次发现的WS相关新变异。
PAX3基因突变导致的WS表型多样且具有异质性。本文4个显性遗传家系的10名患者中,8名为WS1,2名有轻微的手指关节异常;1名疑似新生突变患者为WS3,存在手指弯曲及肘关节突出。Song[13]等报道,PAX3突变患者中50%存在听力损失,本研究收集家系主要因先证者耳聋进行门诊咨询,家系患者中听力异常比例较高,4个显性遗传家系中基因检测阳性者共10人,其中8人(80%,8/10)具有单侧或双侧感音神经性聋,且多为先天性极重度听力损失;虹膜色素异常、內眦异位及额白发亦为常见症状。对于皮肤色素异常,Magnolia[3]等在19例PAX3突变相关WS巴西患者中观察到6例有皮肤白斑,Clinton[4]等也有报道,而中国人群相关报道较少发现[10],本研究患者也未观察到皮肤色素异常,提示皮肤色素异常在中国人群可能较少发生。另外,家系中患者存在外显率的差异。如家系1(图1)中,不同患者听力损失及虹膜色素异常的表现不同;部分家系WS外显率差异较大[14],表型轻微的常被患者忽视,如家系5(图4B)中,I-1內眦间距宽、并眉的表型被其忽视,提示对WS家系成员问诊与查体需要全面,同时应注意遗传学的验证。
PAX3同一氨基酸的改变可造成不同表型。本研究患者2为WS3,检出p.Asn47Lys,与Sommer在CDHS家系中检出的突变相同[6];另外,Sheffer曾在相同位置检出p.Asn47His,表现为WS3[7]。三个家系患者表型有相似也有不同。本家系3中检出的突变为p.Gln264*,表现为WS3,与Regina等在巴西WS1患者中报道的一例新生突变一致[3]。家系4,5的无义突变p.Arg222*在美国家系及中国散发病例中均有报道[4,5],可能是WS较常见的突变位点之一,表型也不尽相同。由此可见PAX3同一位置密码子的相同及不同改变均可造成表型的多样性,可能是氨基酸的不同改变对下游基因及修饰基因等可造成不同的影响,且环境因素对表型也有影响,具体仍需进一步探究。
PAX3突变导致的WS目前尚无有效药物进行治疗,面部畸形及耳聋可行手术干预。上述CDHS家庭中患者通过手术改善了鼻部外形、矫正了手指偏曲[15]。听力损失可能是由于黑素细胞异常导致耳蜗血管纹功能异常最终导致感音神经性聋,可采取人工耳蜗植入进行干预[16,17]。本研究对WS患者进行电话随访时,多数家属表示患者虹膜异色但视力未发现异常,对正常生活无较大影响,而听力问题亟待解决。本研究中,单侧听力损失的患者未行相关医疗干预;3名双侧听力损失患者中,家系4先证者于13个月时行单侧人工耳蜗植入,现7岁,效果较好;家系5先证者单侧人工耳蜗植入时间为3岁,现4.5岁,效果欠佳;家系2患者未予干预,现已成年,只能使用手语交流。可见对于WS双侧听力损失者,应尽早诊断与干预,因听力损失多为极重度,人工耳蜗植入可能是有效手段;单侧听力损失者,如患耳存在残余听力,需关注患侧听力变化,同时更应注意保护健侧耳。
PAX3基因突变导致的WS以显性遗传为主,也存在新生突变,且表型具有一定异质性,同家系成员间外显率也不同,需要临床遗传咨询予以注意。对于有WS家族史的家系应扩大家族成员样本采集,进行全面的问诊和查体,主要诊断标准和次要诊断标准均需注意,同时需要对先证者及家庭相关成员行基因检测,明确分子病因,评估再发风险;对于否定家族史患者,应当对父母进行验证,明确是否为新生变异,以更好进行遗传咨询与指导。
