国家标准GB/T 21316—2007测定动物源性食品中磺胺类药物残留量的不确定度评定
2020-04-20陈章捷
陈章捷


摘要 对国家标准GB/T 21316—2007测定动物源性食品中磺胺类药物残留量的测量不确定度进行评定。根据《测量不确定度评定与表示》(JJF 1059.1—2012)和《化学分析中不确定度的评估指南》(CNAS-GL006:2019)中的规定,建立测定鳗鱼肉中磺胺残留量测定的数学模型,根据测量不确定度的来源,以磺胺甲基嘧啶为例,对各个不确定度分量进行评定和分析。评定结果表明,当样品磺胺甲基嘧啶含量检测结果为10.4 μg/kg时,扩展不确定度为3.4 μg/kg。不确定度按照从大到小排列,依次为标准曲线最小二乘法拟合、测量重复性、回收率、标准溶液制备、LC/MS/MS定量重复性、样品定容、样品称量。
关键词 国家标准;GB/T 21316—2007;动物源性食品;磺胺;不确定度评定
中图分类号 S859.84 文献标识码 A
Abstract The uncertainty of determination of sulfonamide residues in animal-derived foods by GB/T 21316-2007 was evaluated.According to "Evaluation and Expression of Uncertainty in Measurement"(JJF 1059.1-2012)and "Guidance on Quantifying Uncertainty in Chemical Analysis"(CNAS-GL006:2019),a mathematical model for determination of sulfonamide residues in eel meat was established.Based on the sources of the measurement uncertainty,each uncertainty of sulfamethazine as an example was evaluated and analyzed.The evaluation results showed that the extended uncertainty was 3.4 μg/kg when the test result of sulfamethazine content in samples was 10.4 μg/kg.The uncertainty was arranged in order from large to small,which were least squares calibration,measurement repeatability,recovery rate,standard solution preparation,LC/MS/MS quantitative repeatability,sample volume determination and sample weighing.
Key words national standard;GB/T 21316-2007;animal-derived food;sulfonamide;uncertainty evaluation
磺胺类药物是以对位氨基苯磺酰胺(简称磺胺)为基本结构的一类人工合成抗生素的总称,具有抗菌谱广、吸收迅速、使用方便、价格低廉等优点,广泛应用于人类医学和畜牧行业。然而,磺胺类药物在动物体中使用也会产生药物残留风险。这类药物会造成排尿、造血紊乱等副作用和过敏反应,人类若大量摄入将对健康造成危害[1-7]。我国农业部235号公告明确规定,在动物肌肉、脂肪、肝、腎及牛羊奶中,磺胺类药物残留总量不得超过100 μg/kg。国际食品法典委员会、美国、日本、欧盟均对食品中磺胺类药物规定了最高残留限量[1-3,6-7]。
《动物源性食品中磺胺类药物残留量的测定 液相色谱-质谱/质谱法》(GB/T 21316—2007)[8]是国家食品安全监督抽检指定的动物源性食品中磺胺类药物检测国家标准。由于监督抽检结果关乎被抽检的食品生产经营者切身利益,需要对检测结果的不确定度开展评定,以评价抽检结果的准确度。采用该国家标准对市售鳗鱼样品进行磺胺残留量测定,然后依据《测量不确定度评定与表示》(JJF 1059.1—2012)[9]的基本要求,并参考《化学分析中不确定度的评估指南》(CNAS-GL006:2019)[10](该指南等同采用EURACHEM发布的《Quantifying Uncertainty in Analytical Measurement》[11])的评估方法,对检测过程进行了不确定度评定,以期为食品监督抽检机构和检测人员在检测磺胺类药物残留过程中提供技术参考。
1 材料与方法
1.1 主要仪器与试剂
XEVO TQ-S超高效液相色谱质谱联用仪(美国Waters公司);XSE204电子天平(瑞士梅特勒-托利多集团);Milli-Q IQ 7000超纯水机(美国默克集团);移液器(20~200、100~1 000 μL,美国艾本德公司;CR22N高速冷冻离心机(日本日立集团);RE80旋转蒸发仪(日本雅马拓科学公司);KQ-1500DE超声波清洗仪(昆山舒美超声仪器有限公司)。
100 μg/mL磺胺类标准物质(GB/T 21316规定的23种,德国Pharma公司);乙腈、正己烷、正丙醇、甲酸(均为色谱纯,赛默飞世尔科技公司)。
1.2 样品处理
将鳗鱼制成糜样,称取2 g置于50 mL离心管,加入6 g C18填料,使样品与填料混合均匀,加入25 mL乙腈-水(1 000∶30)溶液旋涡振荡1 min,微波炉光波模式下辐照30 s,3 000 r/min离心5 min,将乙腈层移入100 mL棕色分液漏斗,离心后的沉淀物加25 mL乙腈提取30 s,3 000 r/min离心5 min,合并乙腈提取液。在提取液中加入25 mL乙腈饱和正己烷,振摇5 min,将底层乙腈移入150 mL鸡心瓶中,加入10 mL正丙醇,用旋转蒸发仪于45 ℃减压蒸发近干,氮气流吹干。加入1 mL乙腈水溶液(1∶1),超声30 s溶解残渣,将溶解液移入10 mL离心管,加0.5 mL乙腈饱和正己烷,振荡2 min,3 000 r/min离心5min,弃去正己烷,取底层乙腈水溶液过0.22 μm滤膜后,供超高效液相色谱质谱联用仪测定。
1.3 色谱条件
色谱柱:ACQUITY UPLC@BEH C18(2.1 mm×50 mm,1.7 μm);流动相:A相为乙腈,B相为0.1%甲酸。梯度洗脱程序为0~0.2 min,2%A;0.2~3.0 min,2%A加至30%A;3.0~4.0 min,30%A加至70% A;4.0~6.0 min,70%A加至95%A;6.0~6.1 min,95%A减至2%A;6.1~7.0 min,2%A;柱温25 ℃;进样量1 μL;流速0.2 mL/min。
1.4 质谱条件
电离模式为ESI+;监测方式为多重反应监测(MRM);电喷雾电压5 500 V;雾化气压力0.065 MPa;气帘气压力0.016 MPa;辅助气压力0.060 MPa;离子源温度475 ℃。定性离子对、定量离子对、碰撞电压和去簇电压参照GB/T 21316—2007中质谱测定参考条件设置。
1.5 测定
用空白基质配备标准溶液系列,浓度为20、40、60、80、100 ng/mL。按照1.2~1.4方法对鳗鱼样品进行测定。
1.6 计算公式和数学模型
样品中磺胺含量按公式计算:
式中:x为样品中磺胺药物含量单位为μg/kg;c为从标准工作曲线得到的磺胺类药物的浓度,单位为ng/mL;V为定容体积,单位为mL;m为样品质量,单位为g。
上述计算公式是从测量原理给出的,未考虑各种随机因素对不确定度的影响,在此引入反映随机影响的重复性系数frep。另外,由于样品前处理过程较为复杂,通过同时测定加标回收率,引入回收率值R用于校正结果。评定不确定度的数学模型应写成如下形式:
1.7 识别和分析不确定度来源
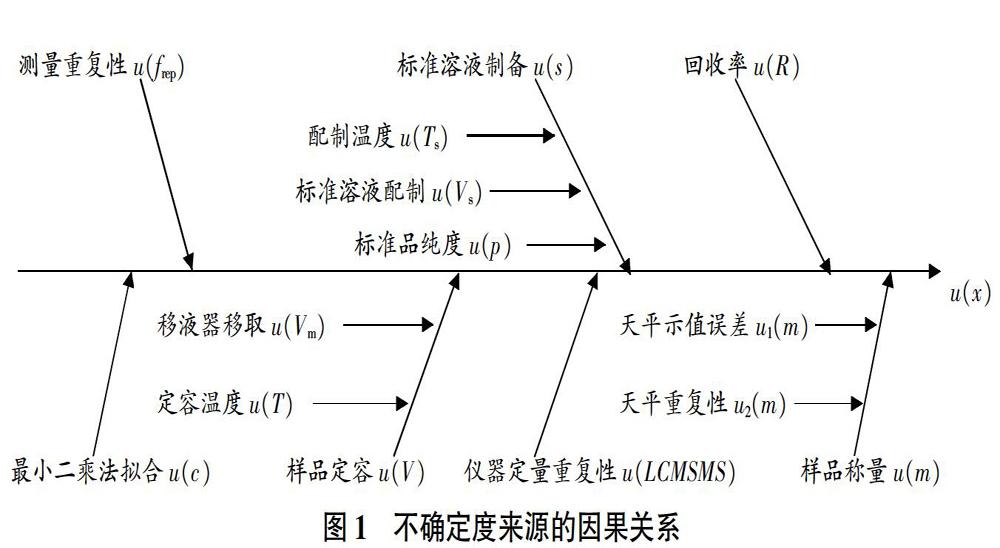
通过绘制因果图来分析农药定量过程中的不确定度来源[12-15],见图1。
2 结果与分析
2.1 不确定度分量的计算(以磺胺甲基嘧啶为例)
2.2 标准溶液制备引入的不确定度
2.2.1 标准品纯度引入的不确定度。磺胺甲基嘧啶标准物质证书中说明其定值为100 μg/mL±3%,则相对扩展不确定度为3%,按正态分布计算,k=2,相对标准不确定度为urel(p)=0.03/2=0.015。
2.2.2 标准中间溶液配制引入的不确定度。用20~200 μL移液器移取标准物质100 μL至10 mL容量瓶,配制1 μg/mL标准中间液。根据《移液器检定规程》(JJG 646—2006),200 μL可调移液器吸取100 μL时容量允许误差为 ±2.0%,按均匀分布,100 μL移液器移取体积误差引入的相对标准不确定度urel(100 μL)=0.012。根据《常用玻璃量器》(JJG 196—2006),A級单标线10 mL容量瓶允差为±0.020 mL,按三角分布计算,包含因子为6,则10 mL容量瓶称量体积误差引入的相对标准不确定度为urel(10 mL)=0.000 82。
2.2.3 系列标准工作液配制引入的不确定度。用100~1 000 μL移液器吸取200、400、600、800、1 000 μL的1 μg/mL标准中间液于10 mL容量瓶,用空白基质配制成系列标准溶液。查《移液器检定规程》(JJG 646—2006),1 000 μL可调移液器移取100 μL时,允差为2.0%,移取500 μL允差为1.0%,移取1 000 μL允差为1.0%。实际移取200、400 μL时允差以2.0%计算,600、800、1 000 μL时允差以1.0%计算。按均匀分布,则urel(200 μL)=0.012,urel(400 μL)=0.012,urel(600 μL)=0.005 8,urel(800 μL)=0.005 8,urel(1 000 μL)=0.005 8。10 mL容量瓶称量体积误差引入的相对标准不确定度urel(10 mL)=0.000 82。
2.2.4 实际操作温度与校正温度不同引入的不确定度。配制过程因与校正温度不同引入的相对不确定度按下式计算:
其中,α为液体的体积膨胀系数,ΔT为温差。
吸取基质为甲醇的母液1次,用乙腈定容1次,吸取基质为乙腈的中间液5次,用空白基质(主要为1∶1乙腈水溶液)定容5次。α甲醇=0.001 18/℃,α乙腈=0.001 37/℃,α水=0.000 208/℃。α乙腈水溶液=0.5α水+0.5α乙腈=0.000 789/℃,ΔT=±3 ℃。urel(T甲醇)=0.002 04,urel(T乙腈)=0.002 37,urel(T乙腈水溶液)=0.001 37。因此,urel(Ts)=0.006 67。
2.2.5 合成标准溶液制备引入的不确定度。
2.3 样品定容引入的不确定度
用100~1 000 μL移液器吸取1 mL乙腈水溶液溶解残渣,移液器允差为1.0%,按均匀分布,则相对标准不确定度urel(Vm)=0.005 8。移液器移取时的温度与校正温度的差异引入的相对标准不确定度urel(T)=0.001 37。因此,样品定容步骤引入的不确定度为urel(V)=0.006 0。
2.4 LC/MS/MS 仪器测定带来的不确定度
计量证书上定量重复性为1%,按均匀分布,仪器定量重复性引入的相对标准不确定度urel(LCMSMS)=0.007 1。
2.5 最小二乘法拟合引入的不确定度
采用空白基质配制5种浓度标液,每个浓度点测定1次,测定数据见表1。通过线性拟合得回归方程y=20 757.730 15x-5.922 04×104,相关系数为0.997 87,方程显著有效(α=5%)。
回归直线的标准偏差按下式计算:
其中,yij为仪器各点响应值;n为测量点数目;m为每个测量点重复测定次数;为回归直线计算的峰面积值。n=5,m=1,计算Sy/x=44 713。
拟合带来的不确定度用下式计算:
其中,B为回归曲线斜率,P为测试样品次数,n为测定标准溶液的次数(总次数),x为未知样的进样浓度,xi为标准曲线每一点的浓度值,x为标准曲线浓度的平均值。计算得u(c)=2.7。
对鳗鱼样品进行一次测定,通过回归曲线计算样品最后进样浓度为20.7 μg/mL,故最小二乘法拟合引入的相对标准不确定度为urel(c)=2.7/20.7=0.13。
2.6 測量重复性带来的不确定度(A类评定)
考虑到同一检测人员在固定的操作环境下严格按照国标方法对样品开展测定,测量系统稳定,根据《测量不确定度评定与表示》(JJF 1059.1—2012),可通过对样品进行10次独立测定,预先评估单个测定值的试验标准偏差。
10次测定结果依次为x1=10.3 μg/kg、x2=11.5 μg/kg、x3=10.5 μg/kg、x4=9.72 μg/kg、x5=9.91 μg/kg、x6=11.6 μg/kg、x7=10.8 μg/kg、x8=9.93 μg/kg、x9=9.79 μg/kg、x10=11.6 μg/kg,平均值x=10.6 μg/kg,测量次数n=10。单次测量的试验标准偏差S(xk)=0.767。
实际测定时只测定1次,n′=1,测定值为10.4 μg/kg,故单次测量值的A类相对不确定度为:
2.7 回收率带来的不确定度
用空白鳗鱼样品以10 μg/kg浓度进行6次加标回收测试,回收率结果见表2。
经计算,平均回收率(R)为89.5%,标准偏差S(R)为0.087 5,平均回收率的相对标准不确定度u(R)=0.035 7。根据《化学分析中不确定度的评估指南》(CNAS-GL006:2019)中偏倚研究的方法,可用t检验来确定平均回收率R是否与100%有显著性差异:检验统计量t=2.93;而95%置信度,n-1自由度下的双侧临界值t(0.05,5)=2.571,t>t(0.05,5),故R与100%有显著性差异,需要进行回收率校正。回收率校正带入的相对标准不确定度urel(R)=0.040。
2.8 合成标准不确定度
试样中磺胺甲基嘧啶含量x的合成标准不确定度计算如下:
3 结论与讨论
依据国家标准GB/T 21316—2007对鳗鱼肉中磺胺类药物残留量进行测定,并以磺胺甲基嘧啶为例,给出了检测结果的不确定度评定示范,为相关食品安全监督抽检报告的结果提供了可靠性评估依据。另外,通过比较不确定度各个分量的值,发现检测过程各个环节对检测结果的不确定度影响程度不同;影响最大的是标准曲线最小二乘法拟合和测量重复性,其余因素按照从大到小依次为回收率、标准溶液制备、LC/MS/MS定量重复性、样品定容、样品称量。因此,检测人员按该标准进行检测时,可通过增加标准曲线每个浓度点的测定次数、增加标准溶液系列点数和增加实际样品平行测定次数等手段,减少标准曲线线性拟合和测量重复性所引入的不确定度,从而减少最终检测结果的不确定度,提高检测结果的可靠性。
4 参考文献
[1] 吴锁连,康怀彬,李冬姣,等.动物食品中磺胺类药物残留的分析方法[J].农产品加工,2017(20):58-67.
[2] 陈振桂.水产品中磺胺类和氯霉素类兽药残留分析方法研究[D].南昌:南昌大学,2008.
[3] 张元,李伟青,周伟娥,等.食品中磺胺类药物前处理及检测方法研究进展[J].食品科学,2015,36(23):340-346.
[4] 王翔,邓晓军,宋国新,等.食品中磺胺类兽药残留前处理技术的研究进展[J].食品科学,2009,30(7):254-257.
[5] 杨海峰.畜产品中抗生素残留的原因及危害[J].中国动物检疫,2007,24(7):19.
[6] 孙亚奇,郭京超,潘源虎,等.磺胺类药物代谢与残留消除以及检测方法研究进展[J].中国兽药杂志,2019,53(2):66-75.
[7] 刘怡君,边海涛,曲宝成,等.食品中磺胺类药物检测方法研究进展[J].食品安全质量检测学报,2017,8(9):3420-3430.
