基于天然产物的铂类和芳基金属抗癌药物研究进展
2020-04-16吕梦迪张自由王萌萌薛旭玲刘红科
葛 超 吕梦迪 张自由 陈 俊 王萌萌 薛旭玲 钱 勇 刘红科
(南京师范大学化学与材料科学学院,南京 210023)
0 引 言
目前,全球范围内癌症发病率逐年升高,尽管有效的预防措施和药物治疗使癌症患者的存活率有所提高,但是目前对许多类型的癌症仍然缺乏有效的治疗手段。以顺铂、卡铂、奥沙利铂为代表的铂类药物是目前临床治疗癌症最广泛采用的金属药物[1]。一般认为顺铂在细胞内经水解后与核DNA结合而阻止DNA复制,从而杀死肿瘤细胞。然而,顺铂对细胞的“无差别攻击”使癌症病人产生了不良反应。如何降低铂类药物的毒副作用成为抗肿瘤药物研究的重要方向之一[2-4]。具有动力学惰性的铂(Ⅳ)配合物可以被细胞内还原性物种(抗坏血酸和谷胱甘肽等)有效还原,从而产生具有高活性的铂(Ⅱ)复合物进入细胞[5]。到目前为止,一些具有药物活性的基团连接在铂(Ⅳ)轴向位置已成为抗肿瘤前药设计策略之一[6-9],因此,将天然产物修饰在铂(Ⅳ)轴向位置实现2种药物的多功能化应用也是铂类药物发展的重要趋势[10]。
除了铂类抗癌药物之外,在开发新型抗癌药物过程中过渡金属配合物显示出良好的前景。相较于有机物,过渡金属配合物具有多种立体化学构型,动力学稳定,并且金属原子在低氧化态时有亲脂性[11-12]。已经报道了许多具有抗癌活性的过渡金属配合物,包括钌、锇、铱、铑、钛、钒、金和铁配合物[11-16]。其中钌(Ru)具有不同的氧化态(Ru(Ⅱ)、Ru(Ⅲ)、Ru(Ⅳ)),在生理条件下稳定而且结构易于调节,几种低毒并具有肿瘤靶向性的钌配合物正在临床试验中[13]。因此,钌配合物成为最受欢迎的新一代金属化疗药物候选者之一[12]。
20世纪 70年代,Clarke等报道的 [Ru(NH3)5(purine)]3+配合物能抑制人鼻咽癌细胞的DNA和蛋白质合成[17],掀起了钌配合物作为潜在抗癌药物的研究热潮[15,18]。在接下来的10年中,Mestroni等开发了以二甲亚砜和氯为配体的六配位Ru(Ⅱ)配合物,其中cis-和trans-[RuCl2(DMSO)4]在体内外都能与DNA相互作用,并且表现出了良好的抗癌活性[19]。由Keppler等研发的 KP1019(图 1)[13]以及 Mestroni,Alessio等开发的NAMI-A(图1)[20-22]正在临床试验阶段。

图1 KP1019[13]和 NAMI-A[20-22]的 结构Fig.1 Structures of KP1019[13]and NAMI-A[20-22]

图2 芳基金属配合物的“琴凳”构型(M=Ru(Ⅱ)和Os(Ⅱ))[27-30]Fig.2 General structure of“piano-stool” arene-metal complexes[27-30]
此外,Clarke等[17]首次报道了三价钌在体内被还原成二价钌的现象,二价钌再与DNA结合并诱导癌细胞凋亡[23-26]。 Sadler等报道了[M(Ⅱ)(η6-arene)(XY)(Z)]n+(图2,M=Ru(Ⅱ)和Os(Ⅱ),XY为双齿螯合配位体,Z是单齿阴离子配体)类型的芳基金属抗癌配合物[27-30],这些抗癌配合物具有特色的“琴凳”结构。其中金属呈低氧化态,这可能是药物对细胞产生毒性的原因[31]。芳烃配体作为η6-电子的给电子体和π-电子的受体,能够稳定金属离子的低氧化态。螯合配体XY的存在,稳定了整体结构功能和优化金属中心电子的性质,单齿配体Z可以给生物分子提供一个空的配位点。此外,还可通过改变芳烃和“琴凳”腿的结构实现金属配合物多功能的性质以及不同的作用机理[32-33],其中具有高水溶性配体PTA的配合物RAPTA-C显示出了较强的抗侵袭和抗转移能力[34]。
研究表明,针对人卵巢癌细胞系的Ru(Ⅱ)和Os(Ⅱ)芳基配合物显示出良好的细胞毒性,部分配合物的活性与顺铂、卡铂相当。例如,Sadler[35]报道的以乙二胺(en)为螯合配体、氯离子为离去基团的配合物(图3),对人卵巢癌细胞A2780的细胞毒性随着配位芳烃大小的增加而增加。离去基团为溴或碘离子时,对配合物的细胞毒性只有微弱的影响。芳基金属的“琴凳”构型易被修饰,通过与生物活性分子结合可以对其物理化学性质和生物活性进行调控。

图3 [(η6-arene)M(en)Cl]+配合物结构(M=Ru(Ⅱ))[35]Fig.3 Structures of[(η6-arene)M(en)Cl]+complexes(M=Ru(Ⅱ))[35]
本课题组近20年来一直从事芳基金属抗癌配合物合成及抗癌机理研究,在芳基金属抗癌配合物与DNA相互作用、方法学研究和实验探索等方面做了一系列工作,为金属配合物在抗癌理论及研究方法学方面做出了一定的贡献[36-43]。Liu等研究了配合物-DNA作用方式与抗癌活性之间的相互关系,发现了配合物与DNA的作用方式与其抗癌活性密切相关;发现稠环芳烃致癌物、双功能配合物与DNA的作用方式非常类似,首次将抗癌机制与致癌机制联系起来,并初步提出了综合考虑抗癌机制与致癌机制相似性理论雏形,为抗癌化学疗法的研究提供了一条新思路[15],这一工作目前已被430多篇SCI源刊物做了高度评价或正面引用。此外,本课题组还首次发现了第三种DNA嵌入类型——贯穿嵌入,“解开了Ru-tha具有高抗癌活性之谜”[36];发现芳基金属配合物以双功能方式 (配位键合,嵌入作用)与DNA作用[37-38],揭示了芳基金属配合物具有与顺铂不同的抗癌机理。目前,本课题组发现具有反式构象的金属大环配合物可以显著影响超螺旋DNA的结构,并诱导DNA缩合[32];合成了系列单核金属大环化合物,这些化合物具有良好的抗癌活性,并且可以在催化、药物递送等中找到进一步的应用[40];设计合成了具有DNA靶向潜力的抗癌剂刚性双核配合物[42];通过改变配体上的取代基,在一定程度上调控了配合物的抗癌活性[42-43]。
以上研究表明,芳基金属配合物具有生物活性好、结构易于修饰等优点,可通过对其结构的微调来调控配合物的抗肿瘤活性。因此将具有不同生理功能的有机分子引入配合物中将有助于调控金属配合物的生物活性,尽可能地拓展化学空间,实现配合物的功能最大化。
从天然产物中提取抗肿瘤活性成分是目前国内外肿瘤药物开发的热门领域[44-46]。我国作为一个天然产物大国,除了具有巨大的天然产物资源外,还拥有源远流长的天然产物开发和使用历史。天然产物具有多种生物特性,是设计和开发癌症化疗新药的重要来源。天然产物有效地促进了第二代药物的开发,包括长春瑞滨和紫杉醇。人们发现许多天然产物能够诱导人类起始肿瘤细胞的凋亡,用作单独治疗或与金属药物联合治疗都有着广阔的发展前景[12]。由于大多数天然产物存在水溶性差、稳定性差、生物利用率低、用药剂量大和毒副作用等缺点,研究人员设计了基于天然产物的铂类和芳基金属配合物,不仅保留了天然产物本身的生物活性和成药性,而且能够改善天然产物的不足,提高其细胞毒性和对肿瘤的选择性,为新型药物设计提供了更广阔的思路。但到目前为止,仅有为数不多的以天然产物为基础的铂类和芳基金属配合物被报道,本文将对有关研究及此类配合物的作用机理进行较为全面的综述。
1 黄酮类金属配合物
1.1 汉黄芩素铂(Ⅳ)配合物
汉黄芩素(5,7-二羟基-8-甲氧基黄酮,图4)是从黄芩根中分离的黄酮类化合物,在体内外均具有抗炎、抗氧化和抗癌等生物活性,是中国及其他几个亚洲国家广泛使用的中草药之一[47]。值得注意的是,它对正常上皮细胞、外周血和骨髓细胞几乎没有毒性[48]。尽管目前汉黄芩素的具体抗癌机制尚不清楚,但是其作为抗癌药物单独或与其他药物联合使用正在Ⅰ期临床试验阶段。
鉴于汉黄芩素的多种生物学功能,将汉黄芩素与铂(Ⅳ)结合作为前药,以期引入汉黄芩素抗炎特征的同时降低铂(Ⅱ)配合物的毒性。Gou等[49]通过酯基将汉黄芩素连接到铂(Ⅳ)配合物的轴向位置,形成的前药配合物1具有较快的还原速率,并且显示出与顺铂相当的细胞毒性(SGC7019:IC50=(3.8±0.1)μmol·L-1,HCT116:IC50=(9.6 ±0.3) μmol·L-1,PC-3:IC50=(4.8±0.2)μmol·L-1)。进一步研究发现,配合物 1 比汉黄芩素抑制COX-2活性的能力更强,从而具有抗炎作用,并且比顺铂更容易将细胞周期阻滞在S期。同时动力学研究表明,配合物1通过激活p53信号通路促进活性氧(ROS)积累,降低线粒体膜电位,进而诱导细胞凋亡(图4)。这些结果证明,与汉黄芩素配位的铂(Ⅳ)配合物1不但能够通过损伤DNA和抑制COX-2表达等多种途径增强其抗肿瘤作用,而且能够提高汉黄芩素衍生物的抗炎能力。综上所述,此研究证明“一体化”前药是开发多靶点肿瘤治疗和提高铂类药物抗肿瘤活性的有效策略。

图4 配合物1抗癌和抗炎的作用机理[49]Fig.4 Anticancer and anti-inflammatory mechanism of complex 1[49]
1.2 3-羟基黄酮金属配合物
天然黄酮类化合物大多存在于植物、水果和蔬菜中,具有抗氧化、抗炎、抗菌和抗癌等多种生物学活性[50]。已报道的具有抗癌活性的金属-类黄酮配合物通过3-羟基黄酮的3号位羟基和4号位酮基与金属离子配位,O,O-螯合配体可以构建高活性及快速水解的芳基金属配合物[51]。
Hartinger 等[52]合成了一系列 Ru(Ⅱ)(η6-arene)的黄酮醇配合物2~4(图5),它们都表现出了良好的生物活性。通过研究配合物的作用模式,发现它们可以与DNA共价结合,并且能够抑制拓扑异构酶Ⅱα活性。与黄酮醇配体相比,Ru(Ⅱ)(η6-p-cymene)-黄酮醇配合物具有潜在的多靶点特性,是更有效的抑制癌细胞增殖药物。

图5 配合物2~4的分子结构[52]Fig.5 Molecular structures of complexes 2~4[52]
Keppler等[53]合成了黄酮醇衍生的 Os(Ⅱ)(η6-cym)和 Rh(Ⅲ)(η5-Cp*)配合物 5~7(图 6),研究了中心金属对水解稳定性、与生物分子反应动力学性质以及细胞毒性的影响,并与Ru(Ⅱ)类似物进行了比较。Rh(Ⅲ)配合物在水溶液中最稳定并且优先与泛素蛋白结合。 此类配合物在 0.8~50 μmol·L-1范围内都表现出优异的抗癌活性。与核苷三磷酸的反应动力学研究揭示了Rh(Ⅲ)与5′-dATP具有较强的亲和力,而Ru(Ⅱ)和Os(Ⅱ)则优先与5′-dGTP结合。配合物与氨基酸和核苷三磷酸同时孵育,发现Rh(Ⅲ)配合物对氨基酸具有优先选择性。这些结果共同表明该类配合物与蛋白质的结合过程可能是抗癌机制的关键因素,说明黄酮醇是一种具有临床应用前景的O,O-螯合配体。
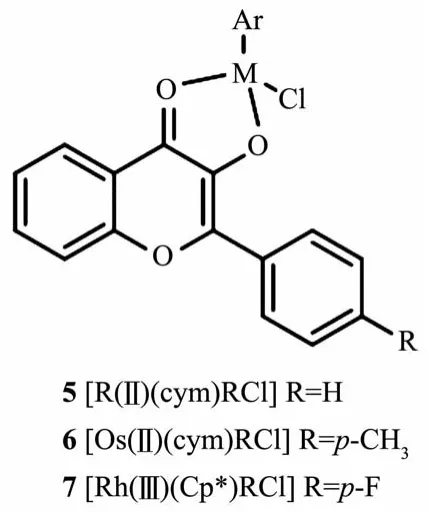
图6 几种黄酮醇衍生的Ru(Ⅱ)、Os(Ⅱ)和Rh(Ⅲ)配合物[53]Fig.6 Several flavonoid-derived Ru(Ⅱ),Os(Ⅱ)and Rh(Ⅲ)complexes[53]
2 β-二元酮金属配合物
2.1 姜黄素铂类金属配合物
姜黄素(Curcumin,curcH)是从姜科植物姜黄中提取的一种天然色素[54],现代研究发现姜黄素具有抗癌、抗炎、抗氧化以及防止动脉硬化等作用[55-56],同时各项动物实验表明姜黄素即使在很高剂量使用的情况下仍然十分安全[57-58]。姜黄素还具有作为抗突变剂和抗癌剂的巨大潜力,并已经进入临床试验Ⅰ期[59],是一种很有开发前景的天然产物。
姜黄素可以通过多种机制发挥其抗肿瘤作用。通过线粒体途径上调Bax蛋白和下调Bcl蛋白,并释放细胞色素C,活化Caspase-9以及下游的Caspase-3引发凋亡[60];抑制 NF-kB信号表达[61-62];调控肿瘤细胞周期[63];抑制肿瘤血管生成[64-65]等。但是姜黄素本身具有水溶性差、稳定性差、生物利用度低以及用药剂量大等缺点[55],限制了其广泛应用。
Chakravarty等[66]合成了姜黄素-顺铂配合物[Pt(curcH)(NH3)2]NO3(8,图7)。它是一种在可见光照射下释放姜黄素和铂(Ⅱ)的新型光动力化疗剂,可以在细胞内同时释放顺铂和姜黄素。与5′GMP和CTDNA的相互作用研究表明,配合物8一旦暴露于可见光(λ=400~700 nm)时可形成 Pt-DNA 加合物。配合物在光照下对肿瘤细胞显示出良好的细胞活性(IC50≈15 μmol·L-1), 而在黑暗中它的活性不会被激活(IC50>200 μmol·L-1)。
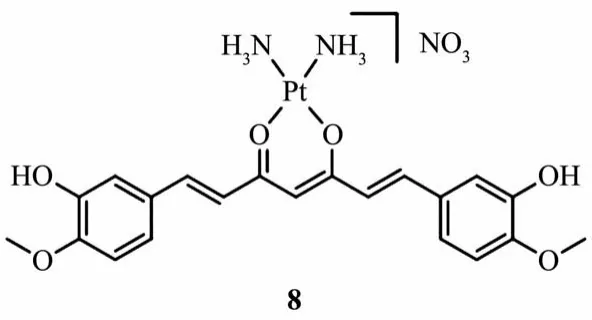
图7 配合物8的化学结构[66]Fig.7 Chemical structure of complex 8[66]
Gamez等[67]报道了配合物 [PtCl(curcH)(DMSO)](9,图8),经研究发现该配合物进入细胞后可释放出游离的姜黄素,姜黄素配体部分对光敏感,形成了一种更有效的抗癌体系,在细胞活性和选择性上都超过了它的母体药物顺铂和姜黄素。

图8 配合物9的化学结构[67]Fig.8 Chemical structure of complex 9[67]
2.2 姜黄素芳基金属配合物
Bonfili等[68]合成了芳基金属钌(Ⅱ)配合物[(η6-arene)Ru(curcH)Cl],芳基为 p-cym(10)、bz(11)和 hmb(12)(图9),并研究了该系列钌配合物调节蛋白酶体的能力。研究发现,3种配合物都可抑制结肠癌细胞中的蛋白酶体,引发细胞凋亡。其中配合物11比10、12和游离姜黄素能更有效地抑制蛋白酶体的活性,具有更好的细胞活性,原因在于它们具有不同的芳烃,可以在不同程度上抑制分离的蛋白水解酶。该工作还研究了配合物对HCT116细胞氧化状态的影响以及配合物11与DNA结合能力,发现配合物11显示出最高的抗氧化能力;此外,与游离姜黄素相比,配合物10和11与DNA有更高的亲和力(高达3 倍)。

图9 姜黄素-钌配合物10~12结构[68]Fig.9 Structures of curcumin-ruthenium complexes[68]
Caruso等[69]合成的姜黄素-对伞花烃钌配合物在结肠-直肠肿瘤细胞系HCT116上显示出优异的活性 (IC50=(13.9±1.5)μmol·L-1), 在乳腺癌 MCF-7(IC50=(19.5±2.3)μmol·L-1) 和卵巢癌 A2780 细胞系(IC50=(23.3±3.3)μmol·L-1)中也显示出良好的抗肿瘤活性,而对成人胶质瘤细胞U-87MG和肺癌细胞A549不敏感。
Dyson等[70]合成了以姜黄素(curcH)和双去甲氧基姜黄素(bdcurcH)为配体的芳基金属钌(Ⅱ)RAPTA型系列配合物 (PTA=1,3,5-三氮杂-7-磷杂三环癸烷),其中配合物[(p-cym)Ru(curH)(PTA)](SO3CF3)(13,图10)对卵巢癌A2780和顺铂耐药株A2780R都具有良好的活性(IC50≤1.0 μmol·L-1),同时配合物对非肿瘤细胞的毒性显著降低。该配合物比[(p-cym)Ru(curcH)Cl]具有更高的抗癌活性,这可能是配体PTA造成的。

图10 配合物13的结构[70]Fig.10 Structure of complex 13[70]
与基于铂或钌的抗癌剂相比,茂基铑、铱和锇的天然产物配合物研究较少。Dyson等[71a]研究了一系列半夹心环戊二烯基铑(Ⅲ)和铱(Ⅲ)配合物[Cp*M(curcH/bdcurcH)Cl]和[Cp*M(curcH/bdcurcH)(PTA)][SO3CF3](M=Rh,Ir)。多数配合物在生理条件下稳定,对人卵巢癌(A2780和A2780R)细胞和非致瘤性人胚胎肾(HEK293)细胞具有中等程度细胞毒性(IC50=10~40 μmol·L-1),但缺乏选择性。 在芳基锇(Ⅱ)配合物[(p-cym)Os(curcH/bdcurcH)Cl]和[(p-cym)Os(curcH/bdcurcH)(PTA)](SO3CF3)中,[(p-cym)Os(bdcurcH)(PTA)]对癌细胞 A2780R(IC50=(2.9±0.2)μmol·L-1)具有较好的选择性[71b]。
3 吡喃酮金属配合物
3.1 麦芽醇铂类金属配合物
吡喃酮通常存在于天然产物中,许多衍生物具有良好的生物相容性和细胞毒性[72]。羟基吡喃酮可以从天然产物中获得,也可以通过不同方法合成,并且易转化为硫代吡喃酮和羟基吡啶酮类似物,它们非常适合药物开发和其他生物应用[73]。其中,麦芽醇是研究最多的化合物之一,它作为一种低细胞毒性的食品添加剂,在某些成神经瘤细胞系中表现出较好的抗肿瘤活性[75]。麦芽醇与金属螯合的配合物已被用于治疗多种疾病[74-75]。
Carland等[76]制备了阳离子配合物[Pt(en)(ma)]+(14)和[Pt(R,R-DACH)(ma)]+(15,R,R-DACH=1R,2R-1,2-diaminocyclohexane,ma=maltolato,图 11)。这些化合物对 HeLa 细胞的毒性约为 32 μmol·L-1,对K562 约为 26 μmol·L-1。 在这些细胞系中,顺铂的细胞毒性比麦芽醇金属配合物高2~3倍,而卡铂的细胞毒性比金属配合物低。

图11 配合物14和15的结构[76]Fig.11 Structures of complexes 14 and 15[76]
3.2 麦芽醇芳基金属配合物
为了开发M(arene)(maltolato)配合物抗癌剂,Sadler等[77]合成了一系列麦芽醇衍生物的配合物。它们在水溶液中快速进行配体交换反应,形成带电荷的水合物[Ru(cym)(L)(H2O)],随后形成二聚体副产物。然而带有乙基麦芽醇配体和2-羟甲基-3-氧代-6-甲基-4(1H)-吡喃配体的配合物更稳定,没有观察到二聚体的形成。尽管有些配合物不易于形成二聚体,但是它们在体外抗癌测定中并没有表现出显著的抗肿瘤活性。
Hartinger等[78]制备了与麦芽醇衍生物配位的芳基金属钌(Ⅱ)配合物(图12),并研究了它们对人源肿瘤细胞系的抗癌活性,发现几种配合物对人肿瘤细胞系SW480和CH1都表现出中等的细胞毒性(IC50=20~60 μmol·L-1),对 A549 细胞的活性较差(IC50>100 μmol·L-1),表明它们具有一定程度的选择性。化合物细胞活性可能与芳基部分的吸电子取代基有关,吸电子基团能够降低IC50值,而给电子基团具有相反作用。此外,还测试了它们与5′-GMP的反应,发现配合物与5′-GMP的反应非常迅速,并且在鸟嘌呤的N7处可以选择性地结合。
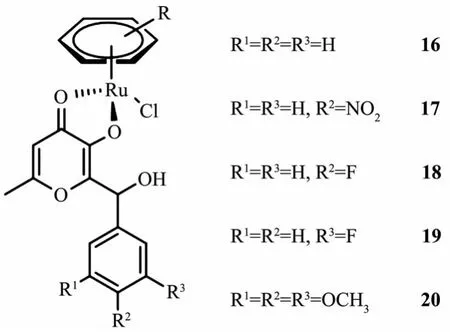
图12 麦芽醇芳基钌配合物16~20[78]Fig.12 Pyrone-based ruthenium(Ⅱ)-arene complexes 16~20[78]
4 β-咔啉金属配合物
β-咔啉生物碱是一类天然存在的化合物,具有良好的生物活性和药理功能,包括镇静作用、抗病毒、抗菌和抗癌活性[79]。据报道,β-咔啉生物碱可通过多种机制发挥抗肿瘤作用,如干扰DNA合成[80],抑制DNA拓扑异构酶Ⅰ、Ⅱ和IκB的活性[81],通过竞争性结合ATP-Mg2+口袋来抑制激酶CDK(细胞周期蛋白依赖性激酶)的活性[82-83]。然而,作为潜在抗癌剂的β-咔啉衍生物的开发受到2个缺点的限制:损伤黑质纹状体多巴胺代谢能力产生神经毒性和有限的水溶性[84]。

图13 β-咔啉配体L1~L3和配合物21~26结构[85]Fig.13 Structures of β-carboline ligands L1~L3 and complexes 21~26[85]
Mao等[85]报道了一系列芳基钌(Ⅱ)配合物(21~26)[(η6-arene)Ru(L)Cl]PF6(芳烃为苯或对伞花烃;L为双齿β-咔啉衍生物)(图13)。这些配合物在体外具有高活性,其中配合物26显示出比顺铂高3~12倍的抗癌活性,且该配合物能够克服顺铂耐药性,同时对正常细胞毒性显著降低。配合物21~26可以将细胞周期阻滞在G2/M期,下调CDK1和细胞周期蛋白B1的表达,同时通过细胞内活性氧(ROS)升高等线粒体相关途径有效诱导细胞凋亡。研究结果说明了抑制CDK1活性是这些配合物发挥抗肿瘤功效的主要机制。芳基钌(Ⅱ)与β-咔啉生物碱的组合是一种有效的策略,可用于开发分子靶向金属抗癌剂,以克服顺铂耐药性。
5 三萜类金属配合物
甘草次酸(18β-glycyrrhetinic acid,GA)大量存在于甘草根中,表现出多样的生物学活性,包括抗病毒、抗过敏、抗炎和抗溃疡作用[86-90]。另外,GA及其衍生物还显示出较好的抗肿瘤活性,例如能够抑制肿瘤的发生和诱导各种癌细胞死亡(包括肝癌、白血病、乳腺癌和胃癌细胞)[91]。GA可直接作用于线粒体,通过线粒体肿胀,线粒体膜电位的丧失以及释放细胞色素C来诱导细胞凋亡[94]。GA也是一种蛋白酶抑制剂,研究显示GA及其衍生物可靶向过氧化物酶激活受体(PPARs)[92-94],并影响肿瘤细胞微环境、血管生成和免疫细胞功能[95-96]。由于GA只表现出中等程度的细胞毒性,因此许多研究人员试图通过GA各种衍生物的合成来提高GA的活性。

图14 甘草次酸、配合物27和28的结构[97]Fig.14 Structures of 18β-glycyrrhetinic acid,complexes 27 and 28[97]
本课题组博士Kong成功合成了2种新型的以GA衍生物为配体的芳基钌(Ⅱ)配合物,并且探索了它们的细胞毒性和选择性(图14)[97]。发现含有N,N-螯合配体的配合物27比含有咪唑作为单齿配体的配合物28具有更好的稳定性和脂溶性。其中,配合物27的水解半衰期是含有2个离去基团(氯化物)的28的3倍,并且27改变了B-DNA的二级结构,在水解时诱导DNA缩合,提升了细胞内ROS水平,破坏了细胞功能,最终导致细胞死亡。曾有报道发现芳基钌(Ⅱ)配合物与2,2′-联吡啶作为二齿配体的配合物是无活性的[33],因此,Kong的结果表明以修饰的GA为配体在调谐芳基金属配合物的化学性质和生物活性中发挥了关键作用。此外,作者还观察到27表现出显著的抗菌活性,可以进一步开发为新的抗菌剂。该工作为天然产物配体修饰及芳基配合物合成打下了良好基础。
6 萘醌类金属配合物
由于植物根部含有大量的白花丹素,所以白花丹叶、石墨玫瑰花和白花丹参都应用于传统医学[98-99]。这种萘醌类化合物具有显著的生物活性,如抗氧化、抗菌、抗炎、抗疟疾和抗癌等作用[100-103]。白花丹素在体外能够诱导拓扑异构酶Ⅱ介导的DNA切割[104]。一些研究通过化学修饰来克服白花丹素的活性和选择性方面的不足,然而醌核心的改变都会减弱白花丹素的活性[102,105]。
为了进一步改善白花丹素的抗癌特性并利用芳基钌配合物的优势,Mahal等[106]将一系列芳基钌连接到白花丹素上(图15),提高了对各种癌细胞系的活性并且改变了作用机理。伞花烃Ru(Ⅱ)配合物29克服了KB-V1/Vbl宫颈癌细胞的多重耐药性,其IC50值约为1 μmol·L-1。此外,还发现针对特定的癌细胞系,天然前体白花丹素及其钌配合物存在不同的作用机制。细胞周期结果发现由白花丹素引起的G2-M期阻滞转变为由配合物29引起的G1-S期阻滞。相较于白花丹素,配合物29很大程度上改变了线性DNA的形状。同时,研究表明,29对多重耐药性KBV1/Vb1细胞的高活性主要是由于配合物对该细胞系中过表达的P-glycoprotein产生了抑制作用。
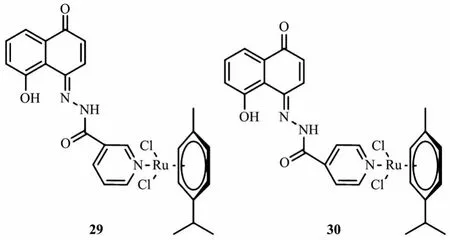
图15 白花丹素-钌配合物29和30的结构[106]Fig.15 Structures of Ru complexes 29 and 30 based on plumbagin[106]
7 研究展望
天然产物具有多种生物特性,用于单独治疗癌症或与金属药物联合治疗都具有广阔的前景,而且某些结构单元是一类理想的先导化合物来源。许多研究表明,以优势结构为基本骨架,通过配位化学等手段,不仅可以保留天然产物的优势,而且可调节铂类及芳基金属的生物活性,达到双功能甚至多功能的效果。因此,借助天然产物的结构先导、修饰和改造合成铂类以及芳基金属配合物,为人们寻找下一代抗肿瘤药物开辟了新思路和途径。此外,目前得到的配合物生物活性大多是体外实验结果,尚未得到体内实验的证实,并且其抗肿瘤作用机制也不明确,因此,基于天然产物的铂类及芳基金属配合物用于生物体内活性研究和作用机理探讨将是未来重要的研究方向。对于这方面的研究本课题组也在深入进行中,希望通过具有不同生理活性的天然产物来增强金属配合物的功能,并研究其相关的作用机制。
