两个Mn(Ⅱ)/Co(Ⅱ)配位聚合物的脲热合成、结构和荧光性质
2020-04-16林星烨杨明星陈丽娟肖芷欣
林星烨 杨明星 陈丽娟 肖芷欣 林 深
(福建师范大学化学与材料学院,福州 350007)
金属-有机配位聚合物在气体存储分离[1-4]、荧光材料[5-6]、磁性材料[7-8]及催化[9-10]等领域具有广泛的应用价值。因此,构筑具有特殊结构及良好性能的金属-有机配位聚合物是化学和材料学的研究热点之一。配合物的结构主要取决于金属离子和有机配体;同时,反应条件如不同的反应温度、pH值、溶液中的抗衡离子以及溶剂的选择和金属离子与有机配体的配比等对于配合物的形成也有重要的影响[11-17]。在有机配体中,多羧酸配体具有很强的配位能力,有灵活多变的配位方式,能与过渡金属离子形成结构多样的配位化合物,在构筑配位聚合物过程中,有着独特的优势,是一类理想配体[18]。4,5-咪唑二羧酸(H3idc)、5-叔丁基间苯二甲酸(H2tbip)含2个羧基,特别是4,5-咪唑二羧酸还含有咪唑基,有着更丰富的配位模式[19]。因此,它们被广泛用作构筑配位聚合物的多齿桥连配体。由H3idc、H2tbip分别与过渡金属在水热条件下已合成得到一系列多维配合物[20-22]。在配合物合成过程中,溶剂的影响不容忽视,尿素或其衍生物类溶剂呈弱碱性,对有机配体有良好的溶解性,有利于金属与配体的配位;同时,它还可以参与配位,作为模板填充在骨架中,当受热时,可能脱去溶剂分子形成孔道结构[23]。因此,以尿素或其衍生物作为溶剂的脲热合成法是获得多孔结构配位聚合物的有效途径之一。迄今为止,以H3idc、H2tbip为配体,以尿素衍生物 1,3-二甲基-2-咪唑啉酮(DMI)和 N,N-二甲基丙烯基脲(DMPU)为溶剂合成过渡金属配合物尚未见报道。因此,我们选择H3idc、H2tbip2个羧酸配体,在DMI、DMPU作为溶剂的条件下,获得了2个含锰或钴的三维/二维金属有机配位聚合物,研究了2个配合物的晶体结构特征和荧光性质。
1 实验部分
1.1 试剂与仪器
H2tbip、H3idc、4,4′-bidpe(4,4′-二咪唑基二苯醚)购于济南恒化科技有限公司产品,MnCl2·4H2O、Co(NO3)2·6H2O、DMI、DMPU 均为分析纯。 红外光谱用NICOLET-5700 FT-IR傅立叶变换红外光谱仪(KBr压片,4 000~400 cm-1)(美国 Nicolet公司)测定。元素分析采用Vario ELⅢ元素分析仪(德国elementar公司)测定。热重分析采用TGA-SDTA851热分析仪,在高纯氮气(载流速度45 mL·min-1)下,以Al2O3粉为参比物,升温速度为10℃·min-1,测试配合物从室温到800℃的热重曲线。粉末XRD采用Rigaku DMAX2500粉末X射线衍射仪,其光源为Cu Kα(λ=0.154 06 nm),工作电压 40 kV,工作电流100 mA,扫描范围 5°~70°(配合物 1)和 5°~65°(配合物2)。荧光光谱则采用FL/FS 920 TCSPC荧光分析仪(358 nm激发)测试。
1.2 配合物的合成
1.2.1 [Mn3(Hidc)3(DMI)2]n的合成
将 H3idc(0.007 8 g,0.05 mmol)、4,4′-bibpe(0.015 1 g,0.05 mmol)、MnCl2·4H2O(0.009 9 g,0.05 mmol)、2 mL DMI和1 mL H2O混合并均匀搅拌10 min,再密封于25 mL的反应釜中,置于程序控温的反应炉中,2 h内升至160℃,并持续3 d,然后以3℃·h-1的速率降到室温。经过滤、洗涤、干燥,得到无色针状晶体,产率约为53%。元素分析值按C25H15Mn3N10O14的计算值(%):C,35.57;H,1.79;N,16.59。实测值(%):C,35.55;H,1.85;N,16.54。IR(KBr压片,cm-1):3 417w,3 133m,2 942m,2 876w,1 672s,1 610s,1 585s,1 527s,1 482s,1 410s,1 388s,1 368s,1 294m,1 255m,1 234s,1 144s,1 112s,1 081s,1 044s,960m,870m,844m,805m,784m,748m,659m,587s,521m,461w,414w。
1.2.2 配合物[Co(tbip)(DMPU)]n的合成
将 H2tbip(0.016 7 g,0.075 mmol)、4,4′-bidpe(0.015 1 g,0.05 mmol)、Co(NO3)2·6H2O(0.029 1 g,0.1 mmol)、2 mL DMI和1 mL H2O混合并均匀搅拌10 min,再密封于25 mL的反应釜中,置于程序控温的反应炉中,2 h内升至160℃,并持续3 d,然后以3℃·h-1的速率降到室温。经过滤、洗涤、干燥,得到绿色块状晶体,产率约为41%。元素分析值按C18H24CoN2O5的计算值(%):C,53.08;H,5.94;N,6.88。实测值(%):C,53.12;H,5.86;N 6.92。IR(KBr压片 ,cm-1):3 413m,2 956m,2 875m,1 646s,1 560s,1 418s,1 382s,1 319m,1 274m,1 070w,942w,920w,782m,751w,719s,488w,435w。
1.3 晶体结构测定
选取大小合适的配合物晶体,在带有石墨单色器的Rigaku 18KWRAXIS-RAPID Weissenberg Imaging Plate衍射仪上进行衍射实验。用Mo Kα射线(λ=0.071 073 nm)和ω扫描方式,分别在2.02°≤θ≤27.48°(1)和 2.32°≤θ≤27.47°(2)的范围内收集到独立衍射点(Rint=0.054 7(1),0.028 6(2)),其中 I>2σ(I)的可观测点用于结构修正。衍射数据用TEXSAN程序进行还原处理,衍射强度经Lp因子和半经验吸收校正。晶体结构由直接法解出,部分非氢原子坐标在随后的差值Fourier合成中陆续确定。对所有非氢原子的坐标和各向异性温度因子进行最小二乘法修正。配合物的所有氢原子为理论加氢,氢原子的坐标和各向同性温度因子参加最小二乘法修正。配合物1采用的权重方案分别为w=[σ2(Fo2)+(0.138 9P)2+0.00P]-1和P=(Fo2+2Fc2)/3,配合物2采用的权重方案分别为 w=[σ2(Fo2)+(0.110 2P)2+1.723 2P]-1和 P=(Fo2+2Fc2)/3,所有的计算均采用SHELXL-2018程序[24]进行。配合物1和2的晶体学参数列于表1,主要的键长键角数据见Supporting information。
CCDC:1943672,1;1943671,2。

表1 配合物1和2的晶体学数据Table 1 Crystal data and structure refinements for complexes 1 and 2
2 结果与讨论
2.1 晶体结构
X射线单晶结构分析表明2个配合物都以尿素衍生物分子为模板剂,由多羧基配体和金属离子构筑的多维金属有机框架结构。有趣的是,实验中加入的4,4′-bidpe配体并未参与配位反应,但未加入4,4′-bidpe 又得不到上述 2 个配合物, 说明 4,4′-bidpe的加入影响了最终配合物的生成。2个体系中都加入了较大量的多羧基配体,由于加入的多羧基配体体积较小且配位能力较强,能迅速与金属离子配位。当羧酸配体与金属离子配位生成稳定的配合物后不利于 4,4′-bidpe 再与金属配位, 因此 4,4′-bidpe在2个体系中可能与多羧基配体共同起到调节pH值的作用。

图1 配合物1的50%椭球率的配位模式图Fig.1 Coordination pattern diagram of complex 1 with 50%probability ellipsoids
配合物1属于单斜晶系,P21/n空间群。如图1所示,配合物1的每个不对称单元含有2个晶体学独立的Mn(Ⅱ)离子,2个占有率为0.5的Mn(Ⅱ)离子,3个配体阴离子Hidc2-和2个溶剂分子DMI。配合物1中Mn(Ⅱ)离子都处于扭曲的八面体配位环境中,其中Mn1、Mn2离子中心均与来自Hidc2-配体的4个羧基氧、2个咪唑氮原子配位,形成六配位[MnO4N2]模式。在Mn1的配位环境中,O3、N5、N8和O3ii这4个原子配位形成赤道平面,O8和O13i这2个羧基氧原子占据轴向位置;在Mn2的配位环境中,4个配位原子O6、N6、N10和O6iv与Mn2配位形成赤道平面,O12和O10iii分别处于轴向位置;在Mn3的配位环境中,位于轴向的2个配位原子为羧基氧原子O7和O7v,位于赤道平面的供体原子分别为O1、O9、O1v和 O9v;在 Mn4 的配位环境中,O2、O14vi、O2vi和O14这4个配位原子处于赤道平面,O11和O11vi这2个羧基氧原子呈轴向分布。Mn-O、Mn-N键长数值与已报道的文献[25]相比,处于正常的键长范围。
配合物1每个不对称单元中的3个Hidc2-配体,分别命名为 L1(含有 O3~O6)、L2 含有(O7~O10)、L3(含有 O11~O14)。L1 为四齿配体,L2、L3 配位模式相同,均为五齿配体。其中L1配体的咪唑基、一个羧基去质子化,这与红外谱图上得出的结果是一致的[26];L1配体的2个羧基都是以一个氧同相邻的咪唑氮与Mn(Ⅱ)离子配位形成五元螯合环(图2)。L2、L3配体的2个羧基均去质子化,并以二齿桥联配位模式与3个Mn(Ⅱ)离子配位。其中一个羧酸的一个氧原子及相邻咪唑氮原子与Mn(Ⅱ)离子构成五元螯合环,另一个氧原子及另一个羧基的氧原子与Mn(Ⅱ)离子配位构成七元螯合环,另一个羧基的另一个氧原子单独和Mn(Ⅱ)离子配位(图3)。
如图4所示,配合物1中L2配体的2个不同羧基上的氧与Mn3螯合,再通过中心对称方式形成Mn(Hidc)2单元;在不同方向上的L3以同样配位方式与Mn4形成另一个Mn(Hidc)2单元;上述2个单元之间再通过Mn1、Mn2连结形成沿a轴无限延伸的一维带状链。
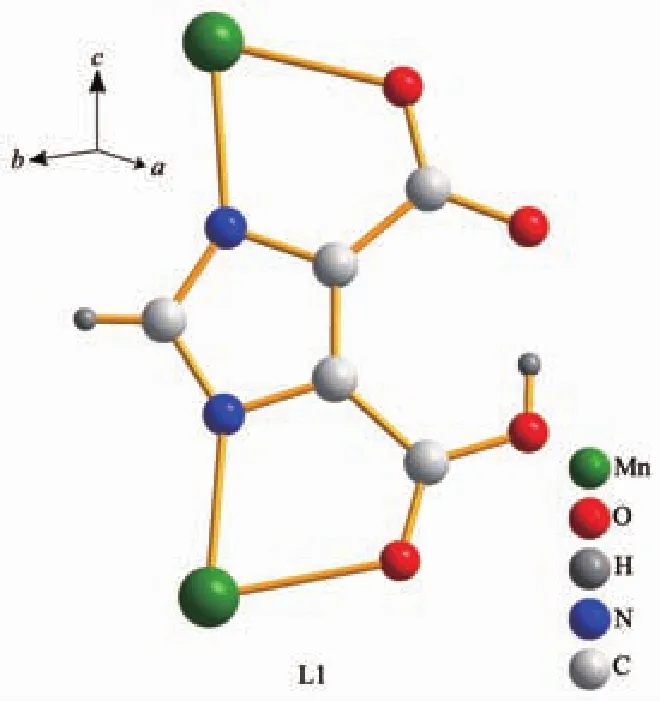
图2 L1配体与金属的连接方式Fig.2 Coordination mode of ligand L1 with metal

图3 L2、L3配体与金属的连接方式Fig.3 Coordination mode of ligands L2 and L3 with metal
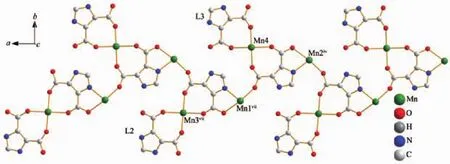
图4 配合物1中的一维带状链结构Fig.4 One dimensional ribbon-like chain structure of 1
如图5(a)所示,一维链中的O13、Mn2分别与另一条链上Mn1、O10配位,形成沿b,c轴方向无限延伸的二维层结构。层与层之间通过L1配体与相邻层中4个Mn原子配位连接形成三维孔道结构(图5(b)),溶剂分子DMI填充在孔道中(图5(c))。其结构与文献报道的以水为溶剂的3个4,5-咪唑二羧酸锰 配 合 物 [Mn(Hidc)(H2O)]n[21]、(Mn(Hidc)(H2O)]n[27]和Mn3(Hidc)2(H2O)4][28]结构完全不同,且文献报道的配合物中配体与金属离子形成的骨架并未形成孔道结构,由此说明了合成条件中溶剂对配合物结构的影响及溶剂分子DMI的模板效应。

图5 (a)配合物1中的二维层状结构;(b)配合物1的三维无溶剂孔道结构;(c)配合物1的三维有溶剂孔道结构Fig.5 (a)Two-dimensional layer structure of 1;(b)3D pore structure of 1 without solvent;(c)3D pore structure of 1 with solvent

图6 配合物1中的氢键示意图Fig.6 Schematic diagram of hydrogen bond in complex 1
如图6(a)所示,配合物1中L2配体内未参与配位的咪唑基和相邻的L3配体中以二齿桥连配位模式的羧基中参与配位的氧原子形成分子间氢键 (图中蓝色虚线);如图6(b)所示,沿c轴方向连接层与层之间的L1配体还存在分子内氢键 (图中粉色虚线)。氢键的存在进一步稳定了配合物的骨架。
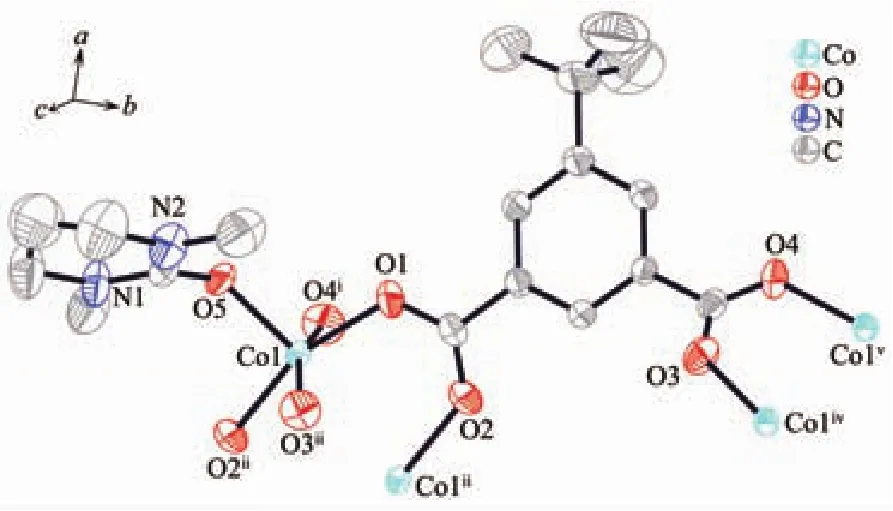
图7 配合物2的50%椭球率的配位模式图Fig.7 Coordination pattern diagram of complex 2 with 50%probability ellipsoids
配合物2属于单斜晶系,P21/c空间群。如图7所示,2的每个不对称单元中含有一个晶体学独立的Co(Ⅱ)离子,一个完全去质子化的tbip2-配体和一个溶剂DMPU分子。中心离子Co(Ⅱ)离子处于三角双锥配位环境中,4个5-叔丁基间苯二甲酸的4个羧基氧及一个DMPU溶剂分子中的酮基氧原子与Co(Ⅱ)离子配位形成CoO5的四方锥配位模式,来自4个H2tbip 配体的 4 个羧基氧原子(O3iii、O1、O4i、O2ii)构成赤道平面;来自DMPU溶剂分子中的酮基氧原子(O5)占据轴向位置。中心金属离子Co(Ⅱ)与氧原子形成的Co-O键的键长处于0.200 1(2)~0.207 4(2)nm之间,与文献[29]报道的Co-O键键长相近。配合物中tbip2-配体作为四齿配体通过羧基基团以二齿桥联配位模式与4个Co(Ⅱ)离子配位。
如图8所示,配合物2中每2个Co(Ⅱ)离子通过4个羧基氧桥联形成一个双核Co单元,该双核Co单元与相邻的4个双核单元通过H2tbip配体桥联形成二维网状结构(图8(a))。溶剂DMPU分子通过酮基氧原子与骨架上的Co(Ⅱ)离子配位填充在网格中(图8(b))。H2tbip与钴离子构筑的配合物中以三维为主[30-34],二维结构的尚未见报道。

图8 配合物2的二维网状结构Fig.8 Two-dimensional layer structure of 2
2.2 TG分析和XRD分析
配合物的热重曲线见图9和10。配合物1在220~300℃之间第一次出现失重,失重率约为12.5%,该温度区间的失重对应于溶剂分子DMI的分解,若DMI完全分解,理论失重率应为27.0%,说明DMI分解不完全。在350℃之后,该配合物开始出现明显的失重现象,表明Hidc2-配体开始进行分解,当温度升至800℃时,总失重率约为66.5%。若最后剩余的为Mn,理论计算值为19.2%;若剩余的为MnO,理论计算值为24.8%。说明当温度达到800℃时Hidc2-配体仍未完全分解。配合物2在300℃以下时,保持相对稳定;在360℃时开始失重,可能对应于溶剂分子及配体tbip2-的分解,最后剩余15.83%;若最后剩余的为Co,理论计算值为14.5%;若剩余的为CoO,理论计算值为18.42%。说明最后剩余的为Co和CoO的混合物。配合物1开始分解的温度为220℃,而配合物2在温度达到360℃时才开始分解。综上可见,配合物2相对配合物1具有更好的热稳定性。

图9 配合物1的热重曲线Fig.9 Thermogravimetric curve of 1

图10 配合物2的热重曲线Fig.10 Thermogravimetric curve of 2
此外,为了验证测试所用配合物的纯度,对2个配合物进行了粉末XRD表征,如图11和12所示,配合物的粉末XRD图与单晶结构模拟图基本吻合,表明所制备的配合物1和2基本为纯相。

图11 配合物1的粉末X射线衍射图Fig.11 Experimental and simulated PXRD patterns of 1

图12 配合物2的粉末X射线衍射图Fig.12 Experimental and simulated PXRD patterns of 2
2.3 荧光性能分析
配合物1在室温下的固态荧光发射光谱图如图13所示。从图中可见,在波长为358 nm的光激发下,配合物在462 nm处有较强的蓝光发射峰,与游离的配体在482 nm处的荧光发射峰相比,配合物的荧光强度大大增强且蓝移了20 nm,可归属为配体的π*-n或π*-π跃迁所引起的荧光发射[35-36]。其荧光强度增强,可能是配体与金属离子配位后有效地增强了配体的刚性,减少了配体内部电荷转移带来的能量损失[36]。配合物不溶于常见的溶剂,因而可作为一种潜在的蓝色荧光材料。

图13 配合物1的室温固态荧光光谱图Fig.13 Solid-state emission spectra at room temperature for 1
3 结 论
选择 H3idc、H2tbip 为主配体,以 4,4′-bidpe 为辅助配体,与过渡金属Mn(Ⅱ)和Co(Ⅱ)盐,在脲热条件下合成了2个以溶剂分子为模板的三维、二维金属有机配位聚合物[Mn3(Hidc)3(DMI)2]n、[Co(tbip)(DMPU)]n。荧光测试结果表明配合物1在可见光区具有发光性能,而且配合物1的溶剂稳定性好,作为发光材料具有潜在的应用前景。
Supporting information is available at http://www.wjhxxb.cn
