氯乙烯SET-DT悬浮聚合动力学和成粒过程的相互关系
2020-04-06韩剑鹏包永忠
韩剑鹏,包永忠
(1 化学工程联合国家重点实验室,浙江大学化学工程与生物工程学院,浙江杭州310027; 2 浙江大学衢州研究院,浙江衢州324000)
引 言
聚氯乙烯(PVC)树脂主要按传统自由基聚合机理进行生产,最主要聚合方法为悬浮聚合[1]。氯乙烯(VC)自由基聚合时,大分子自由基向单体的链转移显著,同时又有自由基移位后的链增长等副反应,导致合成的PVC 含有叔氯和烯丙基等结构缺陷,热稳定性较差[2-3]。随着活性自由基聚合(LRP)技术的发展,很多学者开始研究适用于氯乙烯的LRP方法[4],氮氧稳定自由基聚合[5]、可逆加成-断裂链转移聚合[6]等LRP 方法虽也可用于VC 聚合,但存在聚合温度过高或调控剂制备复杂等不足。Percec等[7-10]提出VC 单电子转移(SET)和单电子转移-蜕化链转移(SET-DT)活性自由基聚合方法,分别以铜/配体或Na2S2O4/NaHCO3为催化体系、碘代烷(如CHI3、CH2I2)为引发剂,在室温附近实现VC 活性自由基聚合,得到的PVC 含有活性碘末端,可进一步调控其他单体的聚合而制备PVC 嵌段共聚物[11-12]。本课题组采用SET-DT方法制备了VC与丙烯酸[13]、VC与丙酰酰胺[14]等亲水单体的嵌段共聚物。
Percec 等[15]针对氯乙烯SET-DT 悬浮聚合速率过低的不足,提出了采用添加相催化剂加速的方法,但相催化剂为胺类阳离子乳化剂,加入过多对PVC 树脂的热稳定性不利。在课题组前期丙烯酸酯的SET-DT 聚合研究中[16],发现降低单体液滴尺寸可以提高聚合速率,说明成粒过程与SET-DT 聚合动力学存在相互关系。由此,预计降低单体液滴尺寸也可提高氯乙烯SET-DT 悬浮聚合速率,有必要研究聚合动力学和成粒过程的相互关系。由于VC悬浮聚合的带压和非均相性,聚合动力学和成粒过程研究多采用离线或平行聚合(同一配方分别聚合不同时间)方法[17-18],操作复杂费时。Brooks 等[19-20]采用在线显微镜观察VC 悬浮聚合中的液-液分散、液滴黏并等过程,并考察了搅拌转速、分散剂种类、引发剂加入方式对成粒过程的影响。Boscher 等[21]采用在线声衰减光谱法研究了PVA 聚合度、浓度和醇解度、搅拌条件对氯乙烯(以氯丁烷为模拟液)在水相分散的影响。此外,也有研究者提出了采用气相色谱法在线测定氯乙烯聚合过程动力学的方法[22-25]。VC悬浮聚合过程中形成的PVC不溶于VC,聚合物在很低转化率就从单体相中沉淀出来,并通过聚并增长为初级粒子[26-27]。由于VC 能部分溶胀于PVC中,聚合同时在两相(含极少溶解聚合物的单体富相和溶胀单体的聚合物富相)中进行,当转化率较高时(70%左右),单体富相消失,聚合仅在聚合物富相中进行[17,28]。
本文利用在5 L 聚合釜上并行建立的在线气相色谱检测和在线粒径分析系统,同时进行聚合动力学和聚合过程中液滴/颗粒粒径分布的分析,进一步结合聚合所得PVC 树脂的形貌观察,考察分散剂种类和浓度、搅拌转速等对聚合速率和液滴/颗粒粒径分布的影响,为阐明氯乙烯SET-DT 悬浮聚合动力学和成粒过程中的相互作用,进而提高聚合速率或降低引发/催化剂用量提供基础。
1 实验材料和方法
1.1 材料
氯乙烯,工业聚合用,纯度大于99.9%,杭州电化集团有限公司提供;去离子水,自制;连二亚硫酸钠(Na2S2O4)、碳酸氢钠(NaHCO3)、碘仿为化学纯,均购于国药集团化学试剂有限公司;甲基纤维素(MC)和羟丙基甲基纤维素(HPMC),日本信越公司产品;聚乙烯醇(PVA,牌号分别为B72和KH20),日本电气化学和合成化学产品;氮气(99.999%)、氢气(99.999%)、正丁烷(分装前99.9%)和高压空气均购于杭州今工特种气体有限公司。
1.2 氯乙烯聚合及分析

图1 聚合釜及测试系统Fig.1 Illustration of autoclave and measurement system
5 L 的聚合反应釜如图1 所示,有温度探头、电磁控制搅拌桨,激光粒度仪PAT 探头以45℃的角度插入反应釜中,距离搅拌桨约2 cm。通过阀和管路将反应釜连接于气相色谱仪的六通阀,于特定时间打开反应釜上的阀取样进行气相色谱分析。
典型的VC 液-液分散和悬浮聚合过程如下:将分散剂、2 L 去离子水和碘仿加入反应釜中,密封反应釜;抽真空,用氮气置换五次。将正丁烷钢瓶与反应釜接通,真空吸入约7 g 正丁烷;再压入约625 g VC 单体;将搅拌转速调整至设定值,升温到35℃,每隔15 min 取样进行色谱分析(GC 型号为安捷伦7820A,PoraBOND U 毛细管柱,FID 检测器,操作参数:阀箱为100℃,六通阀0.01 min 开0.05 min 关,分流比为50∶1,载气(N2)流速为2 ml/min;柱箱温度为100℃下维持1 min,然后以10℃/min 升高到130℃;检测器温度为250℃,H2为45 ml/min,空气为300 ml/min,尾吹气25 ml/min),直至VC/丁烷面积比基本不变;然后将Na2S2O4/NaHCO3水溶液通过氮气压入聚合釜,开始聚合,每隔1 h左右气相取样测量气相组成,通过基于氯乙烯和丁烷在各相分配的模型计算转化率。采用德国Sequip IPAS 粒径分析系统分析单体液滴和聚合物颗粒的粒径分布[选择60 通道测定,由dmin和dmax(分别为每个通道所测的最小粒径和最大粒径)之间的不同粒径粒子的密度分布,计算平均粒径];聚合反应结束后,排除未反应的VC 单体、出料、去离子水洗涤、干燥至恒重,得到“活性”碘封端的PVC(记为I-PVC-I),称重计算转化率。PVC 树脂颗粒形貌通过场发射扫描电子显微镜(SEM,SU-8010,日本日立)观察,喷金75 s。
2 结果与讨论
2.1 氯乙烯SET-DT 活性自由基聚合动力学检测方法的建立
首先通过单独进样确定氯乙烯和正丁烷的出峰位置,再通入氯乙烯和丁烷混合气观察色谱柱的分离效果,结果如图2 所示。可见色谱柱对这两种物质有较好的分离效果,其中峰1 为正丁烷中杂质丙烷。
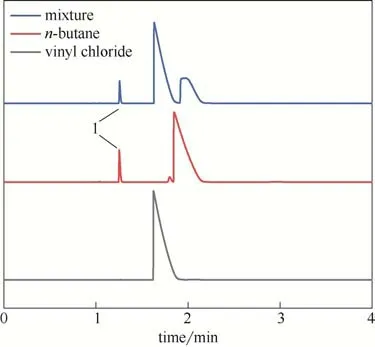
图2 气相色谱对氯乙烯和正丁烷的分离效果Fig.2 Separation of vinyl chloride and n-butane by gas chromatography
氯乙烯聚合转化率可以通过反应t时刻正丁烷的峰面积与t=0 时刻的面积比得到,而无须知道色谱仪器常数和正丁烷总量[22,25]。在搅拌转速为700 r/min、分散剂MC 和HPMC 用量分别为1.4 g 和1.2 g时,进行VC 的SET-DT 聚合,分别采用示踪气相色谱法和称重法测定反应2、4 和8 h 的VC 聚合转化率,得到对比结果如表1所示。可见,通过气相色谱法所得的转化率略大于称重法,但误差不大,这可能是称重法时PVC 样品略有损失所致。尽管氯乙烯SET-DT 聚合过程会产生CO2、SO2等气体[9],但由于气相色谱检测器为FID检测器,不影响测试结果,气相色谱可用于测定VC聚合转化率。


图3 分散剂浓度、种类和搅拌转速对氯乙烯SET-DT活性自由基聚合反应速率的影响Fig.3 Influences of dispersant type and concentration,and agitation rate on SET-DT polymerization rate of VC

表1 不同反应时间气相色谱法与称重法测定氯乙烯转化率的比较Table 1 Comparison of conversion in vinyl chloride polymerization at different reaction time by gas chromatography and weighing method
2.2 分散剂和搅拌转速对聚合反应速率的影响
分散剂和搅拌是影响VC 悬浮聚合成粒过程和PVC 树脂颗粒形态的最主要因素。在引发剂/催化剂浓度不变条件下,改变分散剂种类和浓度、搅拌转速,得到不同条件下的VC 聚合转化率-时间和lnM0/M-时间(M和M0分别为t时刻和t=0 时刻的单体浓度)关系如图3 所示,聚合速率(Kp1和Kp2分别根据lnM0/M-t关系拟合得到)和最终转化率如表2 所示。比较Entry 3 和Entry 4 结果可见,MC 和HPMC分散剂用量增加一倍,Kp1提高近三倍;继续增加MC和HPMC 用量分别为4.2 g 和3.6 g,反应非常迅速,造成反应温度难以控制而发生爆聚。比较Entry 4、Entry 5、Entry 6 发现,反应速率的顺序为:分散剂全为改性纤维素的聚合>改性纤维素/PVA复合的聚合>全为PVA的聚合。
此外,根据速率-t关系可将反应分两个阶段,转变点在转化率50%~70%左右,这与Percec 等[9]的结果相符,但使用全改性纤维素或改性纤维素/PVC复合分散体系的氯乙烯SET-DT 聚合速率明显大于Percec 等[7-9]的结果。两个阶段均近似为一级反应,也证明了聚合的“活性”特征。
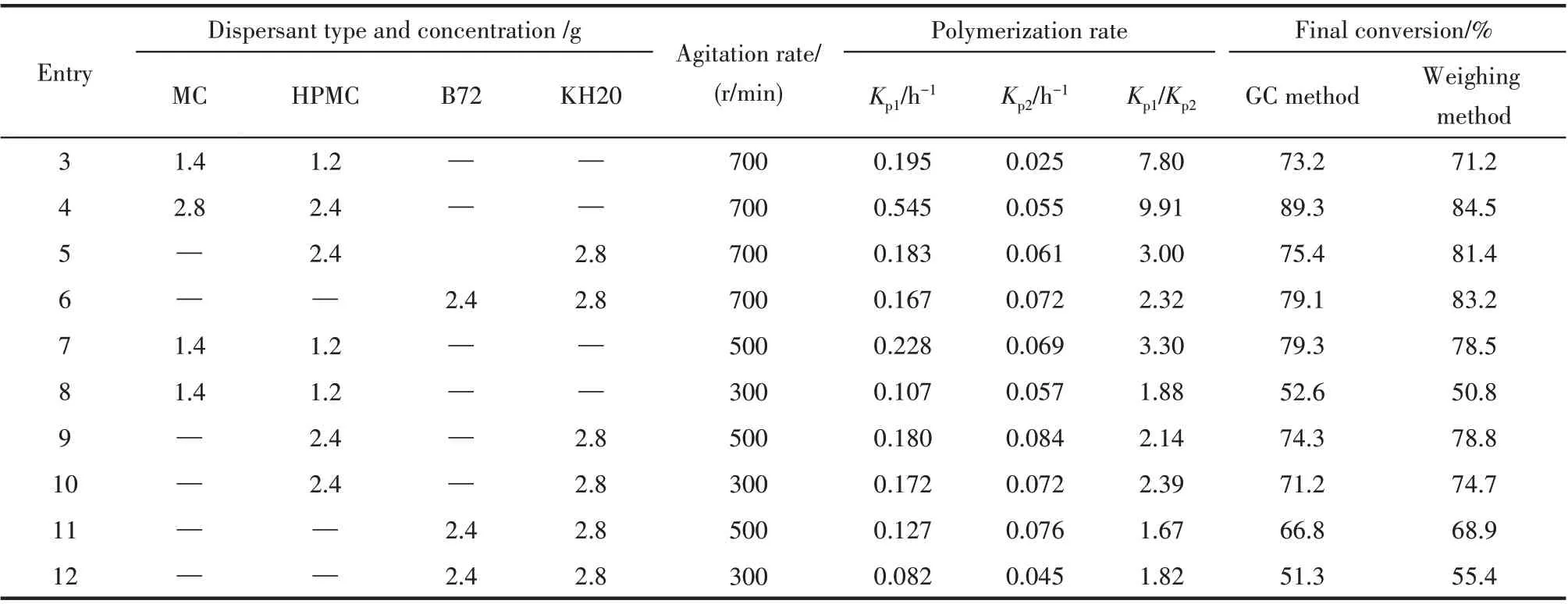
表2 不同分散剂和搅拌条件下的聚合反应速率和转化率Table 2 Polymerization rate and conversion under different dispersants and agitation conditions
图4 和图5 分别为典型的聚合过程中Sauter 平均粒径和粒径分布随反应时间(转化率)的变化。当转化率较低时,液滴的黏度较小,液滴/颗粒破碎速率大于黏并速率,随着聚合时间增加,液滴/颗粒向小粒径方向移动,在30 min 左右达到最小值,此时转化率约为7%;随着转化率继续升高,由于液滴/颗粒黏并速率大于破碎速率,粒径逐渐增大,并在约100 min 时达到平稳,此时转化率为30%左右。

图4 聚合过程Sauter平均粒径随时间的变化Fig.4 Variations of Sauter mean diameter with time in VC suspension polymerization
将横坐标转变为转化率,得到不同分散体系和不同转速下,液滴/颗粒Sauter 平均粒径随转化率的变化如图6所示。尽管分散体系、搅拌转速不同,但VC 的SET-DT 悬浮聚合过程中均可分为三个阶段,且均在转化率为30%左右时进入恒定期。这是由于此时在单体液滴内形成了刚性的聚合物网络以及液滴表面形成皮膜结构(分散体系为PVA 时),这与Kotoulas等[29]的结果一致。

图5 聚合过程粒径分布随时间的变化Fig.5 Time evolution of particle size distribution in VC suspension polymerization

图6 不同分散体系下VC聚合过程液滴/颗粒平均粒径随转化率的变化Fig.6 Evolution of droplet/particle size with conversion of VC polymerization with different dispersant systems

图7 不同分散剂得到的PVC树脂颗粒形貌((a)~(c)MC/HPMC,(d)~(f)HPMC/KH20,(g)~(i)KH20/B72)Fig.7 Morphology of PVC grains prepared by using different dispersants(MC/HPMC(a)—(c),HPMC/KH20(d)—(f),KH20/B72(g)—(i))
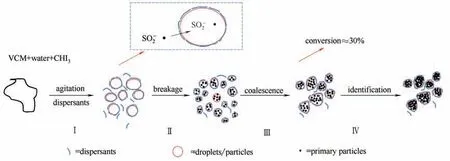
图8 宏观成粒过程Fig.8 Schematic representation of macroscopic particles formation process
不同分散体系下得到的PVC 树脂的SEM 照片如图7 所示。可见,全纤维素分散体系的PVC 树脂颗粒表面无明显的皮膜结构,可以清晰看到初级粒子结构;包含PVA 时颗粒表面有明显的皮膜结构,初级粒子也有明显的皮膜结构,与Zhao 等[30]的观察结果相符。这是由于转化率较低时存在液滴/颗粒破碎黏并,部分分散剂附着在初级粒子表面形成皮膜。
2.3 SET-DT悬浮聚合成粒和动力学的相互关系
根据聚合过程液滴/颗粒分布的变化,提出如图8 所示氯乙烯SET-DT 活性自由基悬浮聚合的宏观成粒过程。


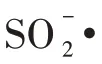
VC悬浮聚合中形成的聚合物不溶于单体,在很低转化率时就沉淀出来,并成长为初级粒子,同时聚合具有单体富相和聚合物富相同时进行的两相聚合特征,聚合从转化率大于30%开始就会出现初级粒子的聚并,进一步降低了自由基由单体相向初级粒子的扩散,因此聚合速率降低。
3 结 论
(1)建立了在线气相色谱法测定氯乙烯SET-DT聚合动力学和在线激光粒度分析测定聚合过程单体液滴/聚合物颗粒粒径分布和平均粒径变化的方法,为研究氯乙烯SET-DT 悬浮聚合动力学和成粒过程相互关系提供了基础。
(2) 相同引发/催化体系下,全改性纤维素分散体系的氯乙烯SET-DT 悬浮聚合具有最大聚合速率,而采用全聚乙烯醇分散体系的氯乙烯SET-DT悬浮聚合速率最小;提高分散剂浓度,可提高聚合速率;分散剂胶体保护充分时,提高搅拌转速也有利于提高聚合速率。
(3) 不同分散体系和搅拌速率下,氯乙烯SETDT悬浮聚合成粒过程均经历液滴分散稳定、黏并和颗粒稳定阶段;采用全改性纤维素分散体系得到的PVC树脂的皮膜结构不明显。
(4) 氯乙烯SET-DT 悬浮聚合动力学与成粒过程存在显著的相互影响,降低单体液滴尺寸和皮膜形成的因素有利于聚合速率的提高,成粒过程中的液滴黏并和初级粒子聚并会导致聚合速率的降低。
