Captiva EMR-Lipid固相萃取/超高效液相色谱-串联质谱法快速筛查动物源食品中51种药物残留
2020-03-19张崇威吴志明李华岑赵逢冰宋志超
张崇威,吴志明,李华岑,赵逢冰,陈 蔷,宋志超
(河南省兽药饲料监察所,河南 郑州 450008)
猪肉、鸡肉及鸡蛋作为人们的重要食物来源,其质量安全问题越来越受到关注。兽药残留作为影响食品安全的重要因素之一,始终是焦点话题,因此对兽药残留的检测显得尤为重要。常用的兽药残留检测方法主要有液相色谱法[1]、液相色谱-质谱联用法[2-4]、气相色谱法[5]、气相色谱-质谱联用法[6]、酶联免疫法[7]等。目前,对动物源食品中兽药多残留检测的研究相对较少,检测方法多为高分辨质谱筛查[8-10],定量效果欠佳。利用液相色谱法[11]及液相色谱-质谱联用法[12-13]进行多残留分析虽有报道,但分析物种类不全、基质单一、操作复杂。例如,郭德华等[14]报道的多残留检测方法不包含氟喹诺酮类、大环内酯类、抗病毒类、镇静剂类及麻醉剂类等常用兽药,且采用HLB柱及MCX柱净化,前处理过程繁琐。郝杰等[15]报道的多残留检测方法不包含硝基咪唑类、镇静剂类及麻醉剂类药物,且检测样品主要为水产品。因此,建立一种快速、简便的猪肉、鸡肉、鸡蛋基质中常用兽药多残留的检测方法显得极为重要。
新型的Captiva EMR-Lipid固相萃取柱可通过体积排阻和疏水相互作用选择性捕获脂类,使体积较大的分析物被阻止在外,因而具有良好的净化效果。本研究以Captiva EMR-Lipid为净化手段,对猪肉、鸡肉、鸡蛋中的β-受体激动剂类、磺胺类、氟喹诺酮类、硝基咪唑类、大环内酯类、抗病毒类、四环素类、抗胆碱类、局部麻醉剂类共9类51种常用药物进行了快速筛查和定量分析。该方法的净化效果好,灵敏度高,且检测药物种类覆盖了目前养殖及运输环节的常用或禁用药物,能够满足猪肉、鸡肉、鸡蛋中多种类兽药筛查确证工作的需要。
1 实验部分
1.1 仪器与设备
Waters Acquity UPLC-Xevo TQ-S三重四极杆串联质谱仪(美国Waters公司);3K-30型台式高速冷冻离心机(德国Sigma公司);TTL-DCⅡ氮吹仪(北京同泰联科技发展有限公司);ES3200型天平(感量0.01 g);Milli-Q超纯水机(美国Millipore公司);KQ 5800 B型超声波清洗器(昆山市超声波仪器有限公司);TARGIN TECH VX-03多管涡旋振荡器(北京踏锦科技有限公司);IKA MS3 Basic圆周振荡器(广州仪科实验室技术有限公司);移液器(德国 Eppendorf 公司)。
1.2 材料与试剂
乙腈(色谱纯,美国Merck公司);甲酸(色谱纯,赛默飞世尔科技(中国)有限公司);Na2HPO4·12H2O、C6H8O7·H2O及Na2EDTA·2H2O(分析纯,天津市科密欧化学试剂有限公司);Mcllvaine-Na2EDTA缓冲液:将12.9 g C6H8O7·H2O、27.6 g Na2HPO4·12H2O和37.2 g Na2EDTA·2H2O溶于900 mL水中,用1 mol/L氢氧化钠溶液调至pH 4.0±0.1,加水稀释至1 000 mL即得。Captiva EMR-Lipid固相萃取柱(3 mL/300 mg,用前无需活化,安捷伦科技有限公司);标准品:51种待测物标准品(见表1),内标物:金刚烷胺-D15、沙丁胺醇-D3、克伦特罗-D9、莱克多巴胺-D3,纯度均大于95%,购自德国Dr.Ehrenstorfer公司。
1.3 标准溶液的配制
混合标准溶液:分别准确称取对照品各约10 mg(以单体计),用甲醇稀释成1 000 μg/mL的贮备液。再分别移取0.1 mL上述各贮备液,以50%乙腈稀释至10 mL,得质量浓度为10 μg/mL的混合标准溶液,于-20 ℃保存。
混合内标溶液:分别准确称取内标对照品各约10 mg(以单体计),用甲醇稀释成1 000 μg/mL的内标贮备液。准确移取0.1 mL上述各贮备液,以50%乙腈稀释至10 mL,得质量浓度为10 μg/mL的混合内标溶液,于-20 ℃保存。
系列标准工作溶液:以50%乙腈将混合标准溶液逐步稀释成质量浓度为5、10、20、50、80、100 μg/L的系列标液中间液。以50%乙腈将混合内标溶液逐步稀释成质量浓度为50 μg/L的内标中间液。分别精密吸取各质量浓度的标液中间液及内标中间液各0.1 mL于相应经前处理所得的空白样品洗脱液中,氮气吹干,以10%甲醇复溶过滤后,待测。
1.4 样品前处理
准确称取猪肉、鸡肉、鸡蛋样品各2 g(精确至0.01 g),置于50 mL离心管中,加入1.0 mL Mcllvaine-Na2EDTA及5.0 mL 0.2%甲酸乙腈溶液,加入陶瓷均质子振荡10 min,于5 ℃以10 000 r/min离心10 min,移取上清液于另一50 mL离心管中,残渣加入4.0 mL 5%甲酸乙腈溶液重复提取1次。合并2次提取液,混匀后于5 ℃以10 000 r/min离心10 min,取上清液2.5 mL过Captiva EMR-Lipid柱,混匀后取2.0 mL滤液氮气吹干,残渣用1.0 mL 10%甲醇复溶,过0.22 μm有机滤膜,上机测定。
1.5 色谱及质谱条件
色谱条件:色谱柱:Waters Acquity BEH C18(2.1 mm×100 mm,1.7 μm);流动相:A为乙腈,B为0.1%甲酸水溶液。梯度洗脱程序: 0~1.0 min,5% A;1.0~3.0 min,5%~15% A;3.0~7.0 min,15%~40% A;7.0~9.0 min,40%~95% A;9.0~12.0 min,95%A;12.0~15.0 min,95%~5% A;柱温:35 ℃;进样量:10 μL;流速:0.3 mL/min。
质谱条件:电喷雾离子源,正离子扫描(ESI+);多反应监测模式(MRM);毛细管电压:1.0 kV;离子源温度:150 ℃;脱溶剂温度:500 ℃;锥孔气流量:150 L/h;脱溶剂气流量:1 000 L/h。51种化合物的定性、定量离子及对应的锥孔电压和碰撞电压如表1所示。
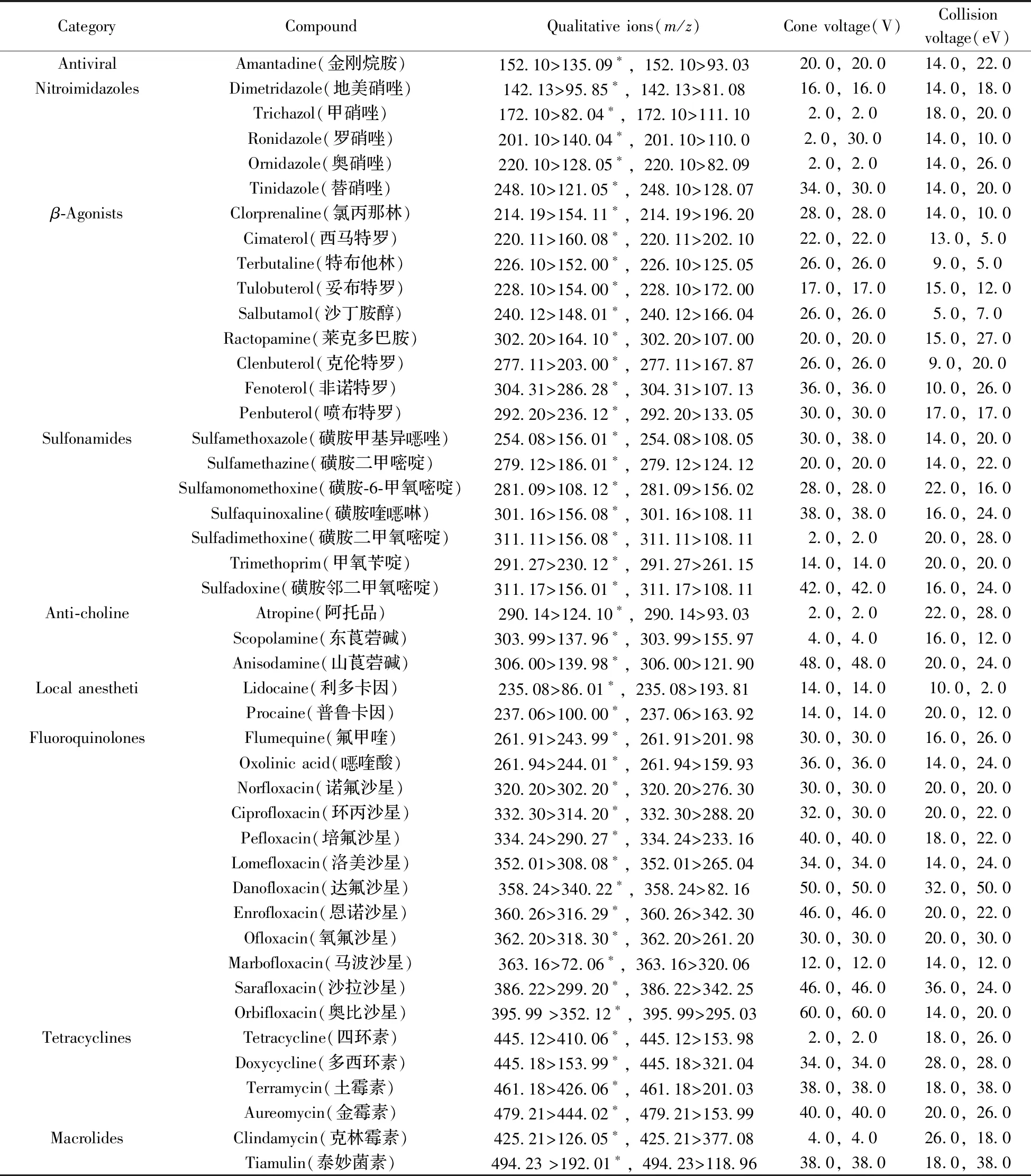
表1 51种化合物的定性、定量离子及对应的锥孔电压和碰撞电压
(续表1)
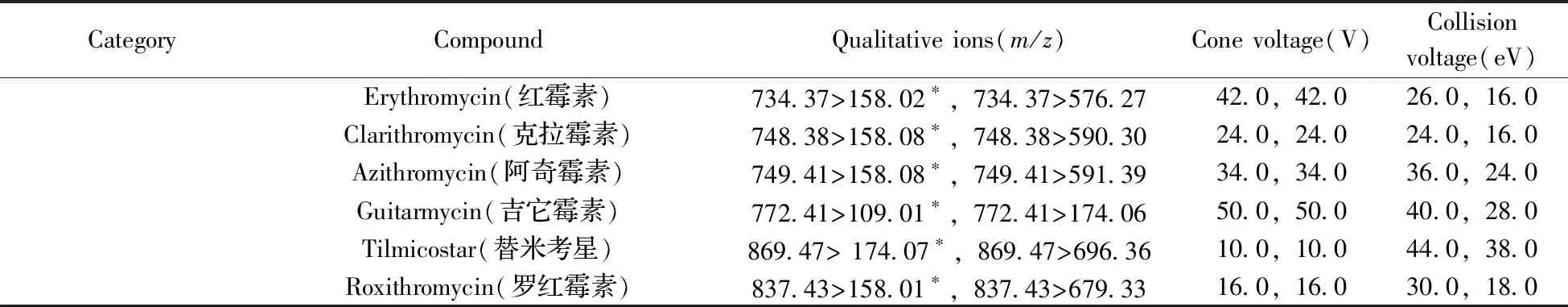
CategoryCompoundQualitative ions(m/z)Cone voltage(V)Collisionvoltage(eV)Erythromycin(红霉素)734.37>158.02∗,734.37>576.2742.0,42.026.0,16.0Clarithromycin(克拉霉素)748.38>158.08∗,748.38>590.3024.0,24.024.0,16.0Azithromycin(阿奇霉素)749.41>158.08∗,749.41>591.3934.0,34.036.0,24.0Guitarmycin(吉它霉素)772.41>109.01∗,772.41>174.0650.0,50.040.0,28.0Tilmicostar(替米考星)869.47> 174.07∗,869.47>696.3610.0,10.044.0,38.0Roxithromycin(罗红霉素)837.43>158.01∗,837.43>679.3316.0,16.030.0,18.0
* quantitative ion
2 结果与讨论
2.1 色谱条件的优化
考察了甲醇-0.1%甲酸水溶液和乙腈-0.1%甲酸水溶液两种流动相体系的分离效果,结果显示乙腈-0.1%甲酸水溶液作为流动相时的分离效果更好,出峰时间更快,缩短了分析时间。进一步比较了乙腈-0.1%甲酸水溶液、乙腈-0.2%甲酸水溶液、乙腈-0.5%甲酸水溶液作为流动相时各化合物的响应值,结果显示响应值无明显变化,考虑到色谱柱损耗及实验成本,最终选择乙腈-0.1%甲酸水溶液作为流动相。并采用梯度洗脱方式,第1.0 min用5%乙腈将极性较大的杂质洗脱,然后逐渐增加乙腈比例,使各化合物在1.0~9.0 min内完全出峰,9.0~12.0 min内乙腈比例保持为95%,将进样通道内剩余杂质全部洗脱,12.0~15.0 min为色谱柱压力平衡时间,为下一针进样做准备,最终实现了51种化合物的有效分离。
2.2 质谱条件的优化
根据文献报道[2-4,12-13],所检各化合物的离子模式均为正离子模式,因此本实验采用正离子模式对各化合物进行全扫描,通过一级质谱获取稳定的[M+H]+分子离子,以确定母离子。MRM模式下,在二级质谱中母离子与Ar碰撞后发生断裂或重排等裂解反应,产生不同质荷比(m/z)的碎片离子,选取干扰最少、强度较高的2个离子碎片分别作为定量和定性离子,并优化碰撞能量使各离子碎片的响应值达到最高。优化的质谱条件如“1.5”所示。
2.3 前处理条件的优化
2.3.1 提取溶剂的选择考察了甲醇、乙腈、乙酸乙酯以及这3种提取溶剂中分别加入0.1%、0.2%、0.5%、1%、5%甲酸或0.1%、0.2%、0.5%、1%、5%氨水对51种化合物提取效率的影响。结果显示,甲醇及乙酸乙酯提取液的杂质多;氟喹诺酮类药物在碱性条件下的回收率很差,只有20%左右。乙腈作为提取溶剂的回收率整体较好,且在乙腈中加入0.2%甲酸可将回收率提高10%以上,这主要是因为待测物均含有碱性基团,甲酸可提供酸性环境,并与待测物形成有机盐,提高待测物极性,根据相似相溶原理,从而提高了待测物的提取效率。随着甲酸比例的增加,大多数化合物的回收率有所提高,但四环素类药物的回收率明显下降,说明该类药物在酸性条件下不稳定。因此,确定第1次提取时采用低浓度的0.2%甲酸乙腈作为提取溶剂,同时加入1.0 mL Mcllvaine-Na2EDTA缓冲液,以提高四环素类药物的提取效率;第2次采用高浓度的5%甲酸乙腈,以增加其余化合物的提取效率。
2.3.2 净化方法的选择目前多残留检测主要采用QuEChERS[16-17]、PEP-2[18-19]、Prime HLB[20]、Captiva EMR-Lipid 4种样品净化方法。对比了上述4种方法的净化效果(如图1),结果显示:QuEChERS法的除杂质效果不理想,基质效应严重,西马特罗、非诺特罗因杂质干扰而无法分辨主峰;采用Prime HLB净化萃取后,四环素类药物的回收率不足50%,可能其对四环素类药物的吸附能力较强,不易洗脱所致;与PEP-2相比,Captiva EMR-Lipid对51种化合物具有更好的净化效果,除四环素类药物的回收率为60%~80%外,其余化合物的回收率均为75%~110%,因此选用Captiva EMR-Lipid作为净化用固相萃取柱。
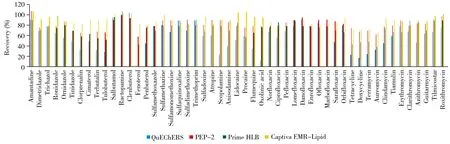
图1 4种净化方法的回收率比较
2.4 基质效应
采用HPLC-MS/MS分析样品时,基质效应是影响定量结果准确性的重要因素[21]。基质效应(ME)是指在样品测定过程中,由于待测物以外其他物质的存在或其他物理、化学因素直接或间接影响离子化效果,从而影响待测物响应的现象。基质效应的计算多采用提取后添加法,该法最早由Matuszewski等[22]提出,是将空白样品处理净化后,在所得空白提取液中添加待测物,在相同仪器条件下,比较空白基质提取液和纯溶剂中目标化合物的离子强度,从而评价基质效应。其计算公式ME(%)=X/Y×100,其中X、Y分别为空白基质提取液和纯溶剂的响应值。若ME值小于100%,表示存在基质抑制效应;若ME值大于100%,表示存在基质增强效应;若ME值等于100%,表示不存在基质效应。表2为部分化合物的基质效应。
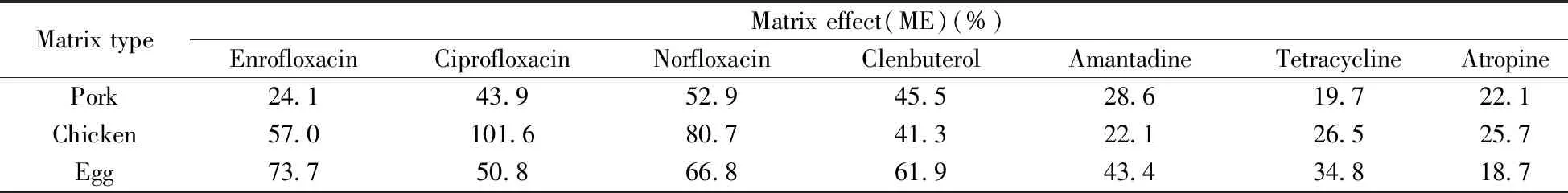
表2 部分药物的基质效应
由表2可知,除鸡肉中环丙沙星外,各目标化合物的ME值均小于100%,表现出基质抑制效应。因此,本实验采用内标及基质匹配方法消除基质效应的影响,沙丁胺醇、克伦特罗、莱克多巴胺、金刚烷胺4种化合物采用内标法进行定量,其余化合物则采用基质匹配外标校正的方法,以确保结果的准确性。
2.5 方法学评价
2.5.1 线性关系对猪肉、鸡肉、鸡蛋中51种药物残留进行检测,采用基质匹配曲线校正,以目标化合物的质量浓度(X,μg/L)为横坐标,相应峰面积(Y)为纵坐标绘制标准曲线。结果表明,猪肉、鸡肉、鸡蛋3种基质中各化合物均在0.5~10 μg/L范围内线性良好,相关系数(r)均不小于0.999 0。其中,猪肉基质的相关参数见表3。
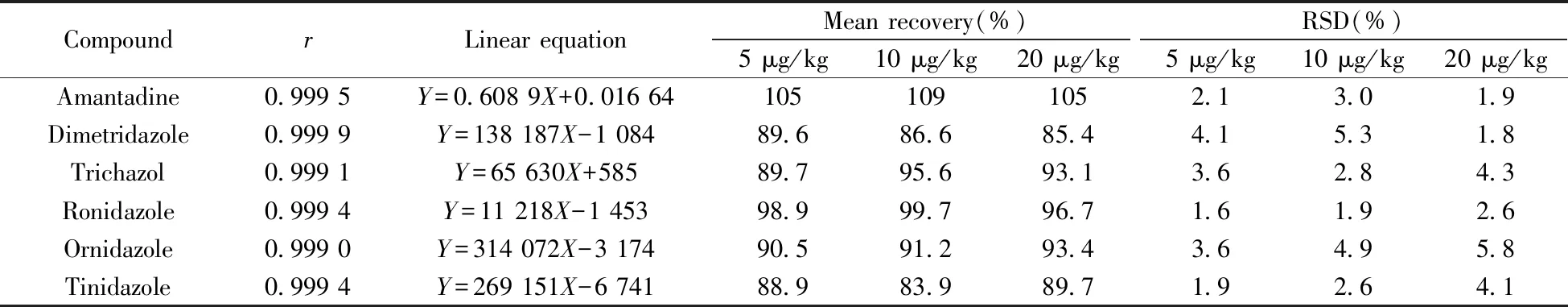
表3 猪肉中51种化合物的相关系数(r)、线性方程、平均回收率及相对标准偏差(n=5)
(续表3)
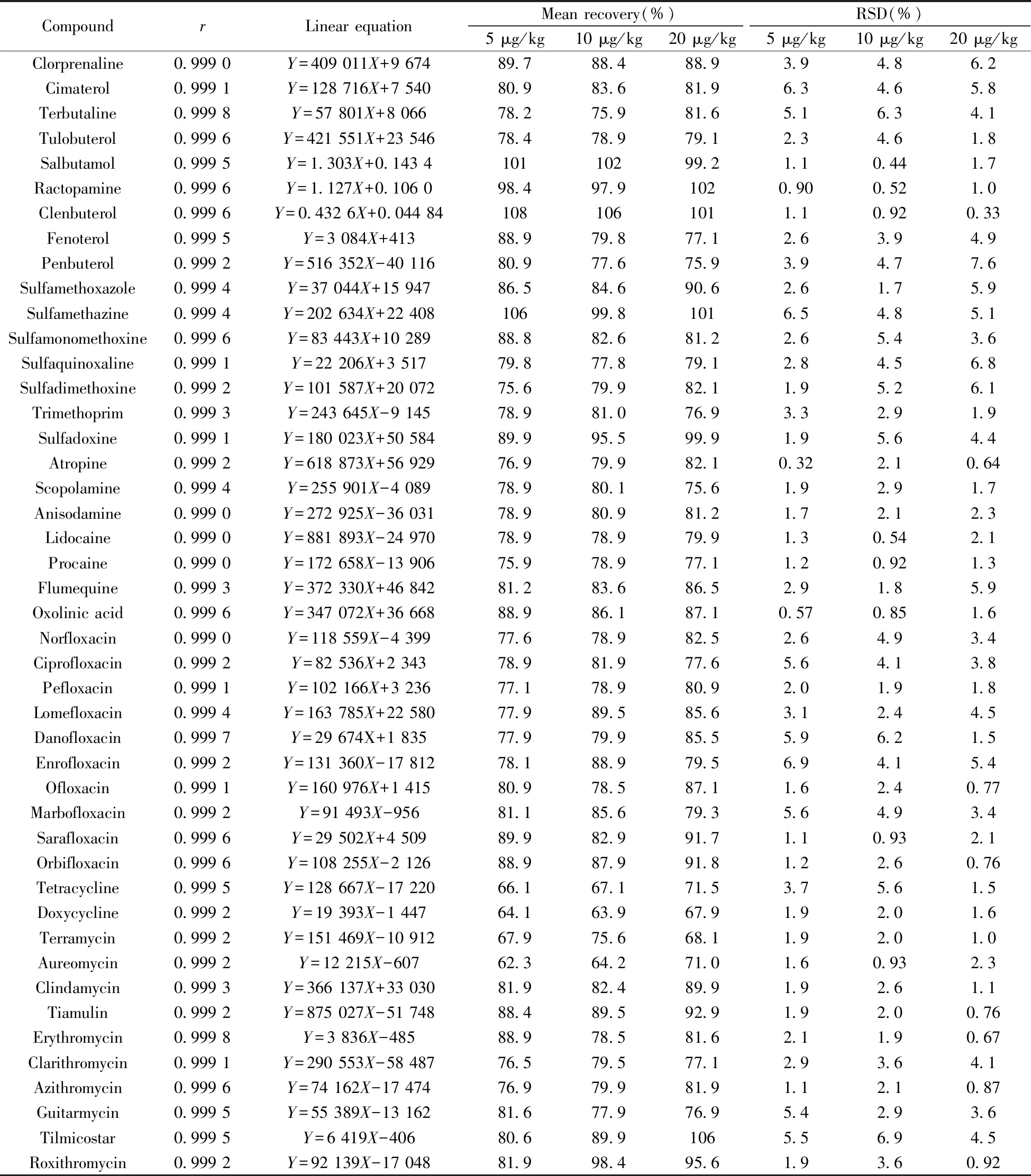
CompoundrLinear equationMean recovery(%)5 μg/kg10 μg/kg20 μg/kgRSD(%)5 μg/kg10 μg/kg20 μg/kgClorprenaline0.999 0Y=409 011X+9 67489.788.488.93.94.86.2Cimaterol0.999 1Y=128 716X+7 54080.983.681.96.34.65.8Terbutaline0.999 8Y=57 801X+8 06678.275.981.65.16.34.1Tulobuterol0.999 6Y=421 551X+23 54678.478.979.12.34.61.8Salbutamol0.999 5Y=1.303X+0.143 410110299.21.10.441.7Ractopamine0.999 6Y=1.127X+0.106 098.497.91020.900.521.0Clenbuterol0.999 6Y=0.432 6X+0.044 841081061011.10.920.33Fenoterol0.999 5Y=3 084X+41388.979.877.12.63.94.9Penbuterol0.999 2Y=516 352X-40 11680.977.675.93.94.77.6Sulfamethoxazole0.999 4Y=37 044X+15 94786.584.690.62.61.75.9Sulfamethazine0.999 4Y=202 634X+22 40810699.81016.54.85.1Sulfamonomethoxine0.999 6Y=83 443X+10 28988.882.681.22.65.43.6Sulfaquinoxaline0.999 1Y=22 206X+3 51779.877.879.12.84.56.8Sulfadimethoxine0.999 2Y=101 587X+20 07275.679.982.11.95.26.1Trimethoprim0.999 3Y=243 645X-9 14578.981.076.93.32.91.9Sulfadoxine0.999 1Y=180 023X+50 58489.995.599.91.95.64.4Atropine0.999 2Y=618 873X+56 92976.979.982.10.322.10.64Scopolamine0.999 4Y=255 901X-4 08978.980.175.61.92.91.7Anisodamine0.999 0Y=272 925X-36 03178.980.981.21.72.12.3Lidocaine0.999 0Y=881 893X-24 97078.978.979.91.30.542.1Procaine0.999 0Y=172 658X-13 90675.978.977.11.20.921.3Flumequine0.999 3Y=372 330X+46 84281.283.686.52.91.85.9Oxolinic acid0.999 6Y=347 072X+36 66888.986.187.10.570.851.6Norfloxacin0.999 0Y=118 559X-4 39977.678.982.52.64.93.4Ciprofloxacin0.999 2Y=82 536X+2 34378.981.977.65.64.13.8Pefloxacin0.999 1Y=102 166X+3 23677.178.980.92.01.91.8Lomefloxacin0.999 4Y=163 785X+22 58077.989.585.63.12.44.5Danofloxacin0.999 7Y=29 674X+1 83577.979.985.55.96.21.5Enrofloxacin0.999 2Y=131 360X-17 81278.188.979.56.94.15.4Ofloxacin0.999 1Y=160 976X+1 41580.978.587.11.62.40.77Marbofloxacin0.999 2Y=91 493X-95681.185.679.35.64.93.4Sarafloxacin0.999 6Y=29 502X+4 50989.982.991.71.10.932.1Orbifloxacin0.999 6Y=108 255X-2 12688.987.991.81.22.60.76Tetracycline0.999 5Y=128 667X-17 22066.167.171.53.75.61.5Doxycycline0.999 2Y=19 393X-1 44764.163.967.91.92.01.6Terramycin0.999 2Y=151 469X-10 91267.975.668.11.92.01.0Aureomycin0.999 2Y=12 215X-60762.364.271.01.60.932.3Clindamycin0.999 3Y=366 137X+33 03081.982.489.91.92.61.1Tiamulin0.999 2Y=875 027X-51 74888.489.592.91.92.00.76Erythromycin0.999 8Y=3 836X-48588.978.581.62.11.90.67Clarithromycin0.999 1Y=290 553X-58 48776.579.577.12.93.64.1Azithromycin0.999 6Y=74 162X-17 47476.979.981.91.12.10.87Guitarmycin0.999 5Y=55 389X-13 16281.677.976.95.42.93.6Tilmicostar0.999 5Y=6 419X-40680.689.91065.56.94.5Roxithromycin0.999 2Y=92 139X-17 04881.998.495.61.93.60.92
2.5.2 检出限与定量下限向空白猪肉、鸡肉、鸡蛋样品中添加一定量的混合标准溶液,按照“1.4”处理后采用“1.5”条件进行分析。 以特征质量色谱峰的信噪比S/N>3时对应的加标水平为检出限,S/N>10时对应的加标水平为定量下限,得到猪肉、鸡肉、鸡蛋样品中51种化合物的检出限均为2.5 μg/kg,定量下限均为5.0 μg/kg。
2.5.3 回收率与相对标准偏差在空白猪肉、鸡肉、鸡蛋样品中分别添加5、10、20 μg/kg 3个浓度水平的药物含量进行回收率实验,每一浓度做5个平行,其中猪肉基质的平均回收率及相对标准偏差(RSD)见表3。结果表明:3种基质中各目标物在3个加标水平下的回收率为60%~110%,RSD小于10%,满足残留分析的要求。图1为空白猪肉中加标量均为5 μg/kg时部分化合物的色谱图。
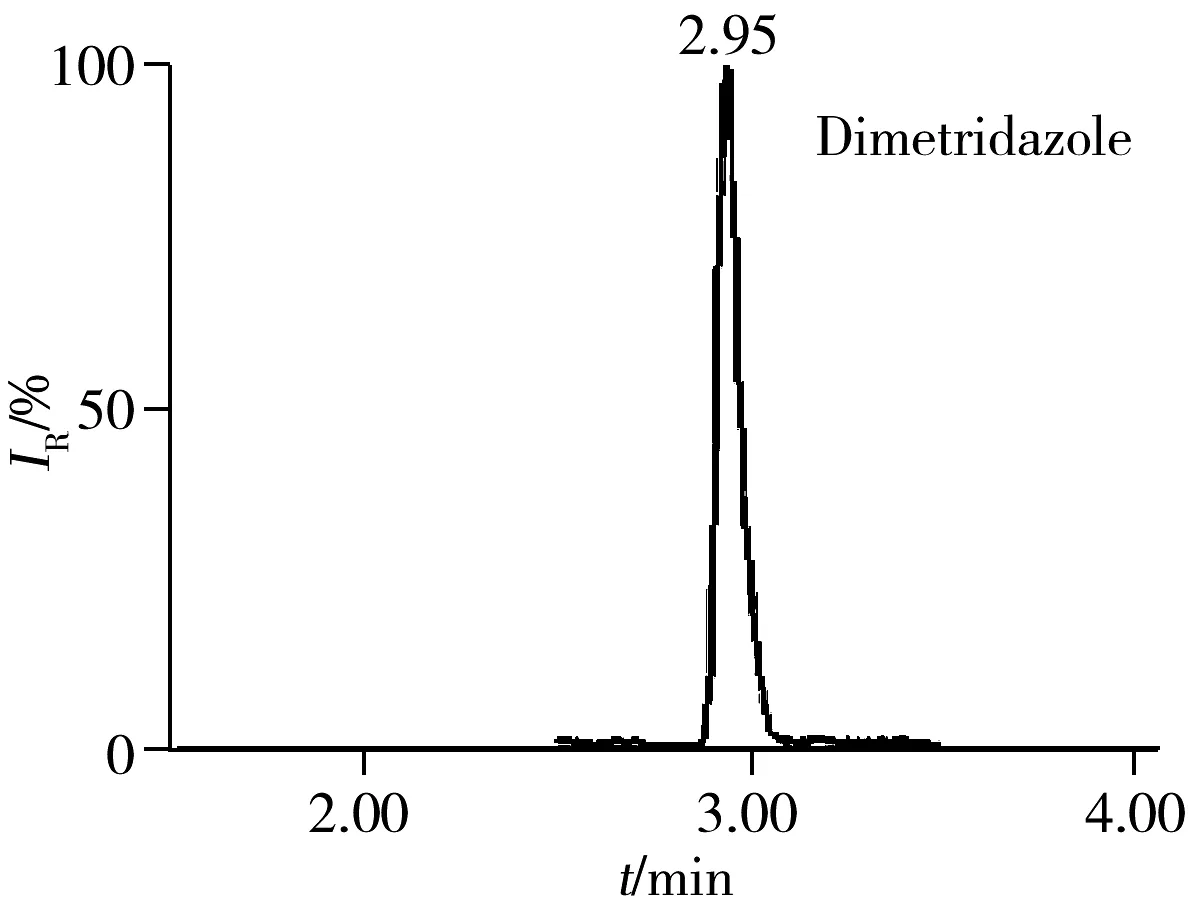
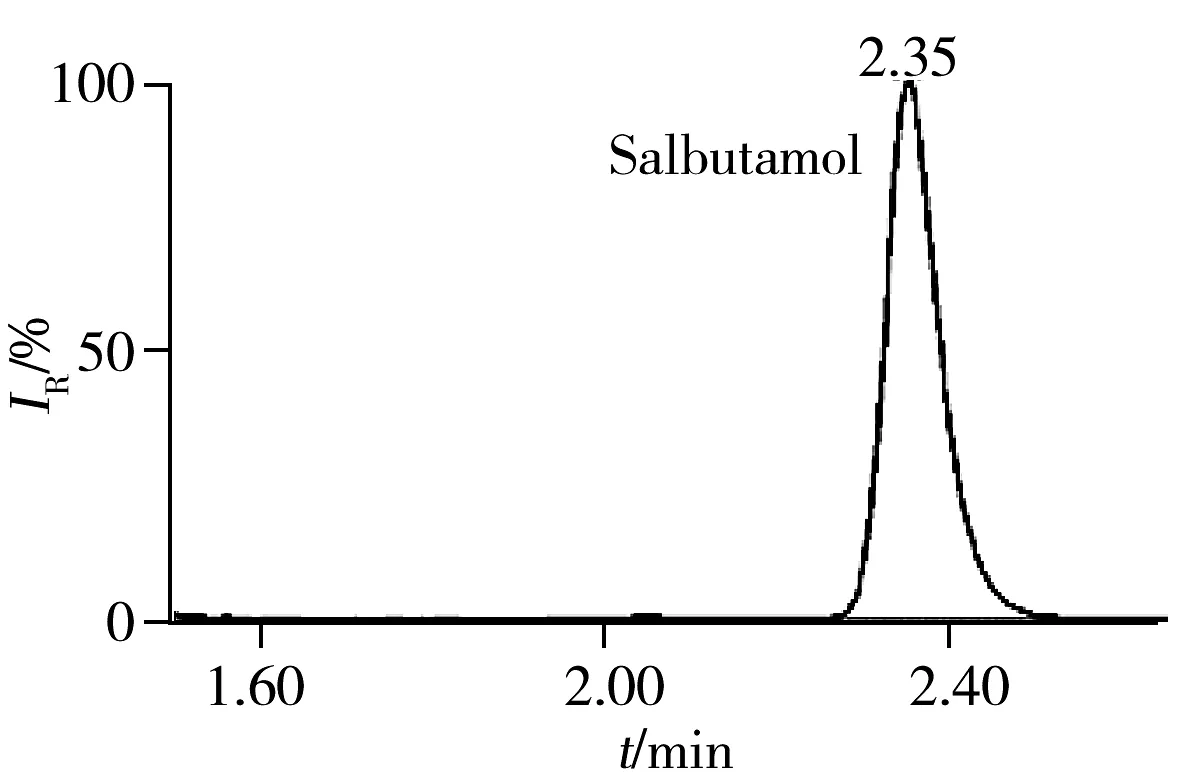
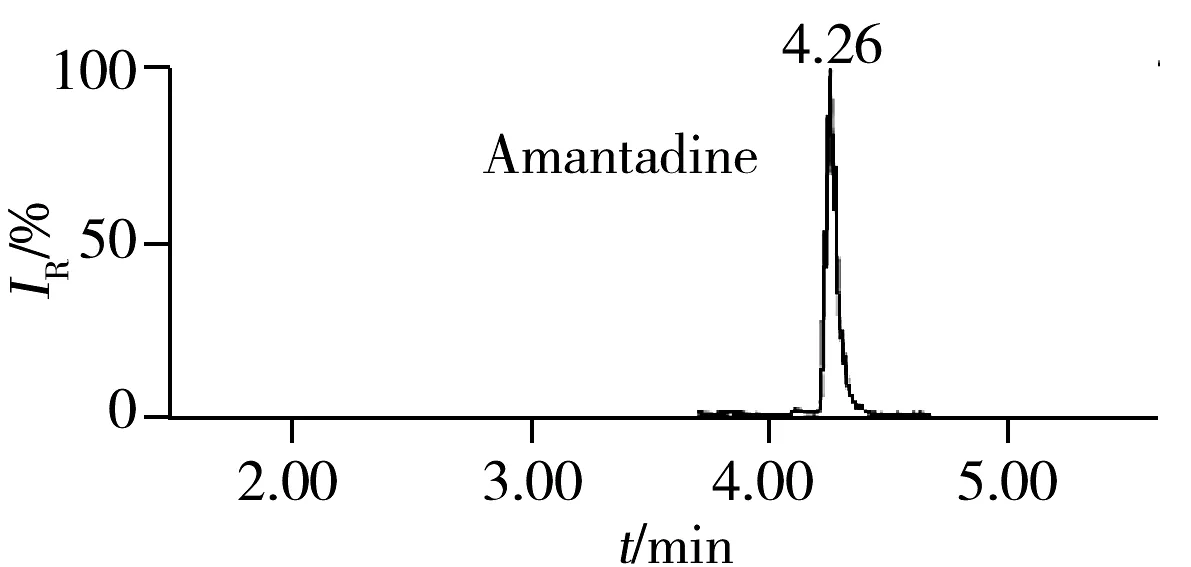
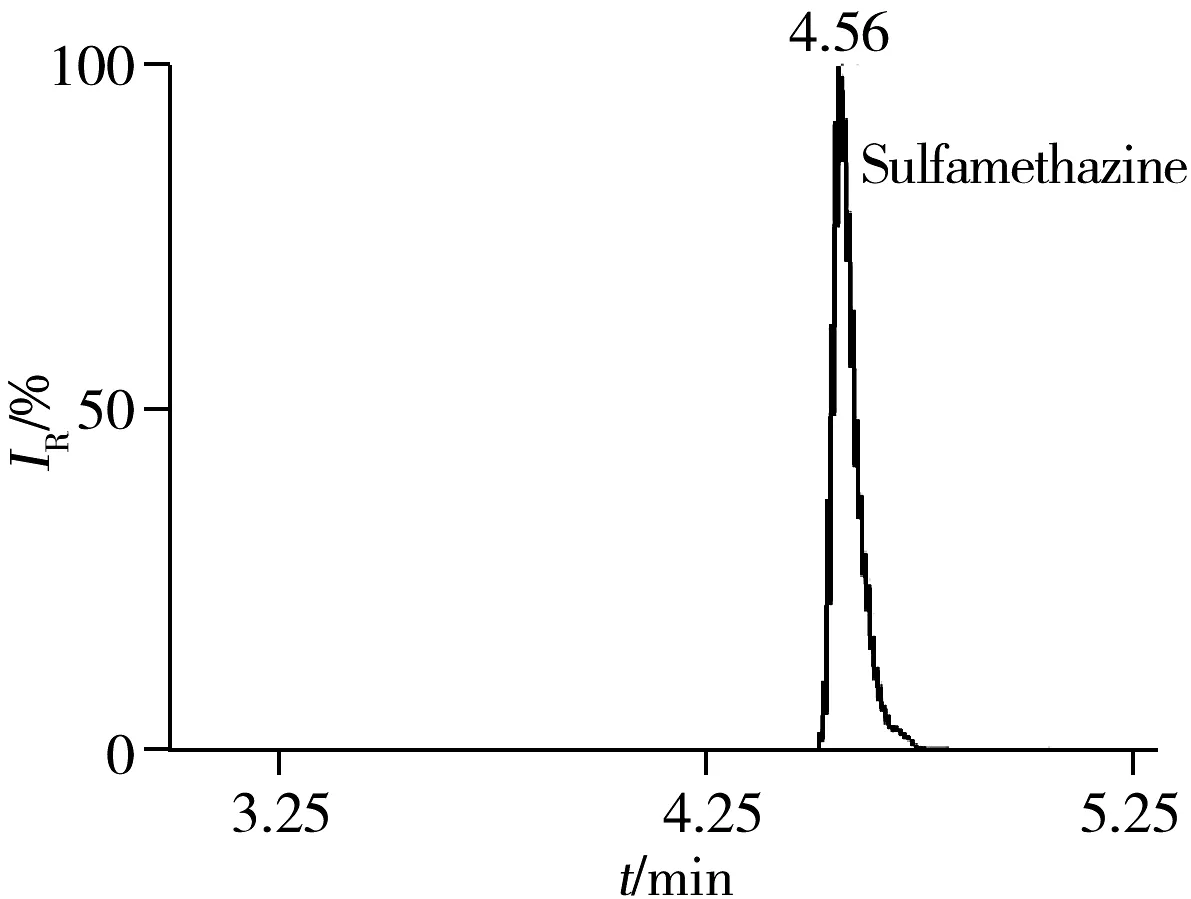
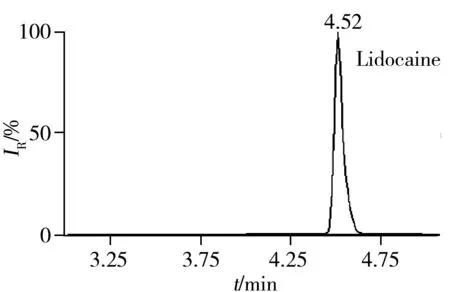
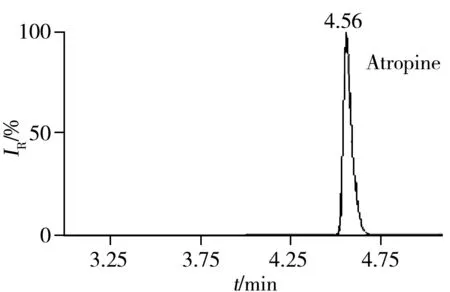
图2 51种药物在空白猪肉基质中添加量为5 μg/kg的代表性药物色谱图
2.6 实际样品的检测
采用本方法对市场抽取的48份猪肉、48份鸡肉和72份鸡蛋进行了检测,同时与国标方法[23-26]相比较。结果显示,2份猪肉中检出多西环素,残留量分别为9.76 μg/kg/11.1 μg/kg(表示本方法检测量/国标方法检测量,下同)、7.44 μg/kg/8.01 μg/kg;5份猪肉中检出金霉素,本方法与国际方法检测的残留量均为5~10 μg/kg。2份鸡肉中检出金刚烷胺,残留量分别为4.90 μg/kg/5.12 μg/kg、3.55 μg/kg/3.81 μg/kg;10份鸡肉中检出土霉素,残留量为5.83~12.9 μg/kg/6.00~14.3 μg/kg;1份鸡肉中检出金霉素,残留量为7.72 μg/kg/7.68 μg/kg。2份鸡蛋中检出金刚烷胺,残留量分别为3.60 μg/kg/3.78 μg/kg、2.74 μg/kg/2.65 μg/kg;2份鸡蛋中检出甲氧苄啶,残留量分别为11.2 μg/kg/10.5 μg/kg、91.3 μg/kg/99.9 μg/kg;1份鸡蛋中检出环丙沙星、4份鸡蛋中检出恩诺沙星、1份鸡蛋中检出氧氟沙星,残留量为3.09~33.9 μg/kg/2.89~48.8 μg/kg;1份鸡蛋中检出多西环素,残留量为25.2 μg/kg/28.4 μg/kg;2份鸡蛋中检出土霉素,残留量分别为46.3 μg/kg/50.4 μg/kg、35.1 μg/kg/33.1 μg/kg。
3 结 论
本研究建立了测定猪肉、鸡肉、鸡蛋中51种药物残留的Captiva EMR-Lipid固相萃取/超高效液相色谱-串联质谱法,各目标物的检出限均为2.5 μg/kg,定量下限均为5.0 μg/kg,在5、10、20 μg/kg加标水平下的平均回收率为60%~110%,RSD小于10%。该方法操作简便、快速、准确度高,能够满足猪肉、鸡肉、鸡蛋中多残留检测的需求。
