酰胺醚类化合物缓蚀行为的理论研究❋
2020-02-06夏树伟张广龙于良民
夏树伟, 张广龙, 于良民
(1.中国海洋大学化学化工学院,山东 青岛 266100;2.中国海洋大学海洋化学理论与工程技术教育部重点实验室,山东 青岛 266100)
金属材料在长期使用过程中会发生腐蚀,造成安全隐患。当前性价比最高的金属防护方法便是添加缓蚀剂[1-4]。但是传统的缓蚀剂设计主要依托高通量的筛选,盲目性大。而通过理论设计,不仅可以深入探究缓蚀剂作用机理,同时还对研发清洁高效的缓蚀剂具有指导作用。
缓蚀剂分子通过自组装,可以在金属表面形成有序的单层膜,有效的减缓金属的腐蚀速率[5-7]。一般而言,缓蚀剂分子都含有N、O等杂原子、不饱和键或共轭结构。酰胺类化合物很好的满足这一结构特点,因此通常被用作缓蚀剂[8-9]。而与之类似的酰胺醚类化合物作为缓蚀剂研究却较少,对此,本课题组已证明该此类物质对碳钢具有缓蚀性能[10-11]。本文选取丙烯酰胺基甲基醚(AAME)[11]及其合成原料—N-羟甲基丙烯酰胺(N-MAM)作为研究对象(缓蚀效率分别为96.2%和93.6%),在分子水平上探索这两种分子的几何结构和电子结构与缓蚀效率的关系;通过分析成键机理,并利用分子动力学模拟探讨了N-MAM与AAME分子在Fe(110)面自组装成膜的行为。
1 理论计算方法
1.1 量子化学计算
应用Gaussian 09[12],选取DFT/M062X[13]方法,分别采用真空和PCM[14]溶剂模型,溶剂的介电常数为78.355 F/m,在6-311+G(d,p)基组下对这两种缓蚀剂分子进行结构优化,并在相同水平下进行频率计算,确保优化至局域最低点。
1.2 分子动力学模拟
选择COMPASS力场,利用Discover模块中的Smart minimizer方法对所构建的模型进行10 000步迭代优化,以避免体系能量陷入低能势阱,在进行能量最小化处理后,模拟温度为298 K,采用Andersen恒温器控制温度,以1 fs为时间步长,进行200 ps正则系综NVT动力学模拟计算。
缓蚀剂分子与Fe(110)面之间的相互作用能Einteraction可通过下式计算:
Einteraction=Etotal-(Einhibitor+Esurface+H2O+H3O++Cl-)。
(1)
式中:Etotal是缓蚀体系的总能量;Einhibitor是单个缓蚀剂分子的能量;Esurface+H2O+H3O++Cl-是Fe(110)面(包括H2O,H3O+,Cl-)的能量。吸附能Eadsorption值与相互作用能Einteraction值互为相反数。
2 结果与讨论
2.1 量子化学计算
缓蚀剂N-MAM与AAME分子在真空和水环境下的优化构型见图1,相关键长、键角总结于表1。相较于真空,水溶液中两种分子的构型发生了一定变化:N-MAM分子的4C-5N和5C-7O键长发生了较大变化,键角∠4N-5C-7O变化最大;AAME分子的3C-4N和9N-10C键长较其它键变化明显,而∠8C-9N-10C是变化最大的键角。由于分子构型发生了一定的变化,所以缓蚀剂分子在真空和溶液环境下吸附到金属表面的构型略有差异。
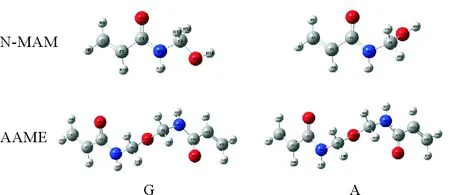
图1 N-MAM和AAME在真空和水溶液中的优化构型(G)真空(A)溶液
两种分子在真空和水溶液中杂原子对最高占据轨道(HOMO)和最低未占据轨道(LUMO)的贡献率见表2。结果表明,N、O原子对HOMO和LUMO的贡献大,N-MAM中N原子在真空和水溶液下对HOMO的贡献率分别达到了53.48%和33.29%,对LUMO的贡献率分别为5.66%和6.03%。而O原子也对HOMO和LUMO具有较大贡献;AAME分子中的N原子在真空和水溶液下对HOMO的贡献率为56.50%和51.08%,对LUMO的贡献率为3.74%和5.26%;O原子对HOMO的贡献率分别为36.41%和13.55%,对LUMO的贡献率分别为10.96%和10.95%。因此可以认为N、O原子是主要活性吸附位点,优先同金属原子发生作用。
根据前线轨道理论[15],分子的反应活性可以通过分子HOMO和LUMO的分布和组成得知。HOMO与LUMO的能级同分子得失电子的能力有关[16]。EHOMO越大,分子供电子能力越强;ELUMO越小,分子接受电子能力强。由表3可知在溶液环境下,AAME的EHOMO值大于N-MAM,即AAME分子供电子的能力强于N-MAM分子。溶剂化作用下,两种分子的ELUMO均比真空中小,说明在水溶液环境中这两种分子的接受电子能力增强。另外,能隙ΔE(ELUMO-EHOMO)能够反映缓蚀剂分子吸附在金属上形成保护层的稳定性。不论在真空或是水溶液环境中,两种缓蚀剂的ΔE相差较小,说明这两种分子吸附到金属上的趋势基本相同,但AAME分子的ΔE小于N-MAM,ΔE越小,分子的反应活性越强[17],分子越容易与金属发生作用,所以AAME分子更易吸附在Fe表面上。
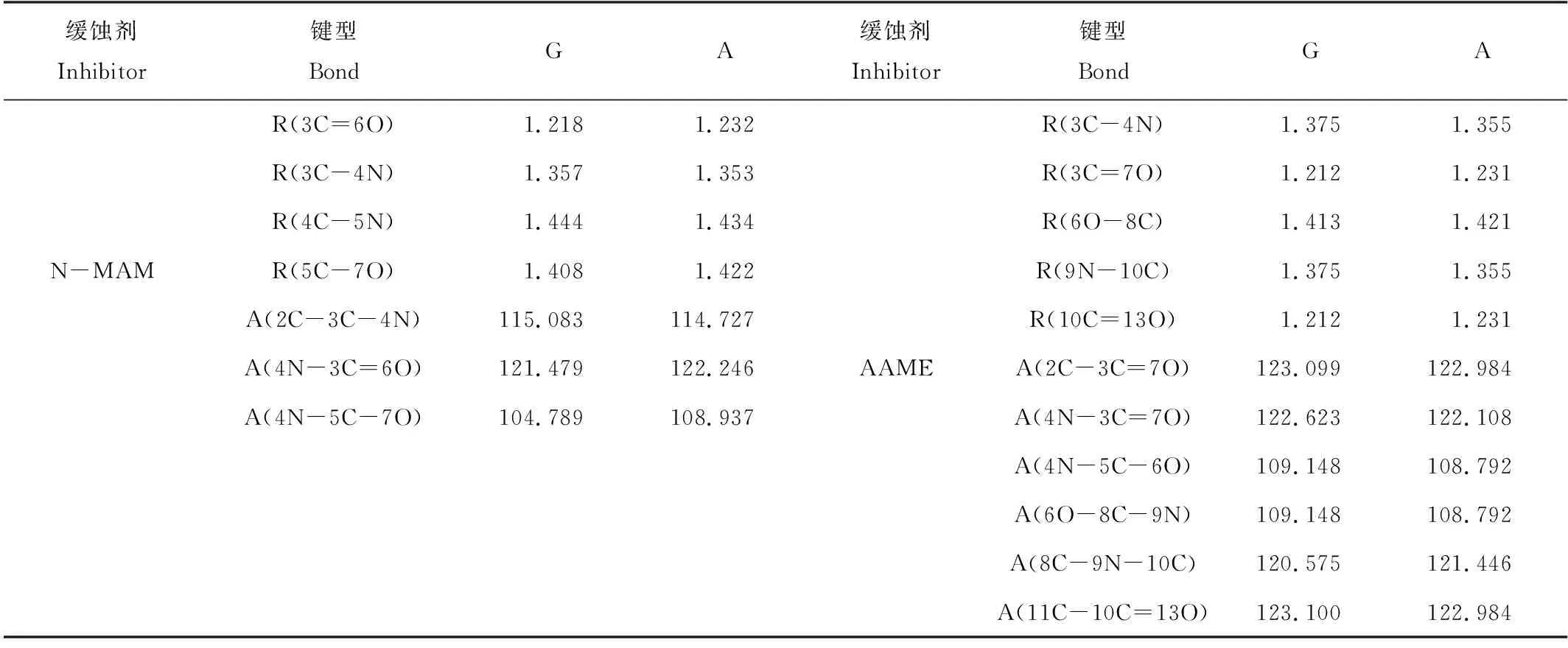
表1 缓蚀剂分子N-MAM与AAME在真空及水溶液中稳定构型结构参数
Note:R:键长 Bond distance/(Å),A: 键角 Bond angle/(°);G-vacuum,A-aqueous phase.
由表2和3可知,在水溶液中分子的总能量(HF)下降,水的溶剂化效应使分子中氧原子的电荷变负。偶极矩(μ)是反映缓蚀剂缓蚀性能的一个重要参数。μ值越高,缓蚀剂在金属表面有强偶极-偶极相互作用[18],会加强缓蚀剂分子在金属表面的吸附性,增强缓蚀效果。溶剂化效应使N-MAM和AAME分子的偶极矩(μ)增大,所以在水溶液中这两种缓蚀剂分子缓蚀性能提高。在缓蚀剂研究体系中,用分子平均极化率〈α〉(〈α〉=αxx+αyy+αzz)来预测缓蚀剂的缓蚀效率。〈α〉越大,缓蚀剂分子在金属表面的吸附就越强[19-20],缓蚀效率越高。溶剂化效应使得两种分子的〈α〉值变大,提高了缓蚀效果。其中,AAME的〈α〉值均大于同等环境下的N-MAM,说明AAME的缓蚀效果更好,与实验所得结果一致。

表2 缓蚀剂N-MAM与AAME分子中原子电荷分布和对前线轨道贡献
Note:G-真空,Vacuum;A-液相,Aqueous phase.

表3 缓蚀剂N-MAM与AAME分子的量子化学参数
Note: G-真空,Vacuum; A-液相,Aqueous phase.
2.2 分子动力学模拟
N-MAM与AAME分子在真空和溶液环境中平衡后的吸附构型分别见图2。对于N-MAM,其在真空和溶液环境下在Fe(110)面吸附的构型相似,仅在分子与Fe(110)上表面的平均距离上有所差异:该平均距离由真空时的2.815 Å,减小至溶液中的2.564 Å,说明N-MAM分子在溶液中向金属表面吸附的趋势变大。而AAME分子在真空和溶液中,分子吸附前后差异较大,说明水的溶剂化效应对AAME分子影响大:AAME分子在真空时距Fe(110)面2.803 Å,而在水溶液中的平衡构型距Fe(110)面2.976 Å。不论在真空还是溶液环境中,缓蚀剂分子都是以近乎平行的方式吸附在Fe(110)面上,所以其对金属具有良好的缓蚀作用。
两种缓蚀剂分子在真空和溶液中的吸附能见表4。两种分子与Fe(110)面的相互作用能均为负值,说明该吸附过程能够自发地发生。相比于真空,在溶液中两种分子在Fe(110)面的吸附能变大,即溶液环境中两种缓蚀剂分子能更稳定地吸附在Fe(110)面,缓蚀效果增强。此外,AAME的吸附能均高于相同条件下的N-MAM,说明AAME效果更强,从而具有更高的缓蚀效率,这与实验所得缓蚀效率顺序一致。

图2 真空和溶液环境中N-MAM和AAME分子在Fe(110)面上的平衡构型

表4 N-MAM和AAME与Fe(110)面的吸附能
2.3 分子自组装成膜的分子动力学研究
分子自组装按其驱动作用大致可分为氢键、配位键、疏水作用、静电相互作用和范德华力等[21],其中氢键作用有利于分子间相互形成网状结构进而成膜。N-MAM与AAME分子中皆含有C=O和N-H结构单元,相邻分子的该类基团之间相互靠近,易形成“C=O…H-N”氢键网络,促使分子自组装成膜。
图3分别为2个N-MAM/AAME分子在Fe(110)面的平衡构型:N-MAM分子间通过羰基上的O原子与相邻分子的N-H、C-H形成“V”型的“C=O…H-N”和“C=O…H-C”氢键。其中,前者的强度较高,对形成氢键网络起主要作用,而后者由于O…H间距离较远,仅起到调整和稳定构型的作用。类似的,AAME分子间通过形成“C=O…H-N”、“C=O…H-C”、“O…H”和“N…H”氢键连接在一起,其中“C=O…H-N”和“C=O…H-C=C”氢键主要为分子内氢键,对形成氢键网络起到主要作用,“O…H”和“N…H”氢键为分子间氢键,主要调整分子在Fe表面的构型。

图3 2个N-MAM(A)与2个AAME分子(B)在Fe (110)面平衡吸附构型
由于这两种分子可以通过氢键作用平铺在Fe表面,因此考虑其成膜性,图4为4个N-MAM分子或3个AAME分子动力学模拟构型。4个N-MAM分子也平铺在Fe(110)面上,且分子间的氢键类型并未改变,仅氢键强度略有下降(见表5)。4个N-MAM分子更有利于形成覆盖度大的氢键网络结构,并在Fe(110)面沿[1 -1 1]晶向依次逆向铺展成膜。3个AAME分子的构型较2个AAME分子变化较大,主要由于AAME分子体积大产生的空间位阻导致的,但分子的疏水基团“C=C”远离Fe表面,这样有利于阻隔腐蚀介质(如H+、H3O+、Cl-)与Fe表面,抑制腐蚀。3个AAME分子相邻分子间氢键强度比2个分子时要强(见表5),形成一个更加稳定的氢键网络结构,并沿[-1 1 0]晶向铺展成膜。因为Fe(110)面是一个周期性结构,所以将Fe晶面周期性放大,可更清晰观察到N-MAM与AAME分子在Fe晶面通过氢键作用铺展成膜,见图5。

图4 4个N-MAM分子(A),3个AAME分子(B)在Fe(110)面平衡吸附构型

表5 不同体系中氢键距离

图5 N-MAM(A)和AAME(B)分子在Fe(110)面通过氢键作用铺展成膜
因为N-MAM/AAME分子可通过氢键作用,在Fe(110)面上能够自组装成膜。对大多数分子而言,色散力是分子间主要的作用力[22]。色散能计算公式如下:
(2)
式中:α是分子极化率,单位为10-40C·m2·V-1;I是分子电离能,单位为kJ·mol-1;ε0是真空电容率,单位为C2·J-1·m-1;R为分子质心距离,单位为10-10m。
分子间的色散能计算结果列于表6。由表可知,N-MAM和AAME分子间平均色散能均较高,说明分子间的作用力较强,有利于分子在金属表面成膜。3个AAME的分子间平均色散能高于4个N-MAM分子的,说明AAME分子更易进行分子自组装成膜,因此缓蚀性能更优。

表6 不同体系中分子间平均色散能
3 结语
采用密度泛函理论(DFT)和分子动力学模拟(MD)等方法探究了N-MAM与AAME分子对碳钢的缓蚀行为,结果表明N-MAM与AAME分子的HOMO和LUMO主要分布在N、O杂原子上,且其对前线轨道贡献率大,表明N、O原子是主要的活性位点。水的溶剂化效应使分子的缓蚀性能比在真空条件下得到了提升。溶剂化效应对AAME分子在金属表面吸附的影响较大,AAME在Fe(110)面的吸附能值均大于相同条件下的N-MAM,表明AAME分子的缓蚀效果优于N-MAM分子。N-MAM和AAME分子均可通过分子间氢键作用相互连接成膜铺展在铁表面上形成一个保护层,能有效地阻隔腐蚀介质(如H+、H3O+、Cl-)减缓铁的腐蚀。
