基于模块度函数的加权蛋白质复合物识别算法
2020-01-17毛伊敏刘银萍
毛伊敏,刘银萍
江西理工大学 信息工程学院,江西 赣州341000
1 引言
在蛋白质相互作用(Protein-Protein Interaction,PPI)网络中,蛋白质复合物是指在相同时间和空间通过相互作用组成一个多分子机制的蛋白质集合[1]。大量的生物实验和计算方法实验产生了许多高质量、大规模的PPI 网络数据,这些数据为识别蛋白质复合物奠定了基础,而蛋白质复合物的识别能够帮助人类预测未知的蛋白质功能,解释特定的生物进程,并为研究疾病的发生机理,寻找新的药物靶标,提供重要的理论基础[2]。因此,识别蛋白质复合物是生物信息领域中的一项研究热点。
迄今为止,早期识别蛋白质复合物的方法是生物实验方法。虽然生物实验方法挖掘复合物的精度较高,但是该方法不仅费时费力,检测效率也已经不能满足后基因组时代的发展。随着高通量生物蛋白信息学的发展,产生了许多高质量和大规模的蛋白质相互作用数据,这些数据为采用计算方法识别蛋白质复合物奠定了基础[3]。因此,出现了大量基于计算方法识别复合物的算法。由于生物网络中每个蛋白质有着不同的功能,不同的边的重要性也不同,为了更真实、详尽地表达出蛋白质网络结构的复杂性和多样性,目前从加权PPI网络中识别蛋白质复合物越来越受到人们的关注[4]。Dimitrakopoulos等人[5]提出一种从加权蛋白质网络之中预测重叠蛋白质复合物的算法GENA(Gradually expanding dense neighborhoods)。基于COACH(Core-attachment method)方法,Kouhsar 等人[6]提出一种快速高性能的预测加权蛋白质复合体检测算法WCOACH。Ama 等人[7]提出一种跨模块中心移除的加权蛋白质复合物识别算法IMHRC(Inter module hub removal clustering)。虽然这些加权网络蛋白质复合物检测算法具有一定的成效,但是由于相互作用数据中存在着较高比例的假阳性和假阴性,以及蛋白质相互作用数据中包含不稳定或发生在不同时间点的相互作用,导致识别蛋白质复合物的精确度不高[8]。为了进一步提高识别的准确性,研究者们开始结合蛋白质相互作用以及多元生物数据来识别复合物。例如,在蛋白质相互作用网络中,胡伟等人[9]提出蛋白质复合物识别ICMDS算法(Predicting complexes based on multiple biological data sources)。该算法整合了基因表达谱、关键蛋白信息和蛋白质相互作用数据。基于蛋白质相互作用数据以及异构生物数据,赵碧海等人[10]提出一种基于多关系网络中关键模块挖掘的蛋白质功能预测算法PEFM(Prediction of protein functions based on essential functional modules mining)。多元生物数据整合能够有效地弥补相互作用网络不完整性和噪声的问题,使得识别蛋白质复合物的精确度得到提高。除此之外,由于模块度函数充分考虑PPI网络的节点分布情况,可以通过优化该函数将连接紧密的节点聚到相同的模块中,符合蛋白质复合物的结构特征,因此研究者提出了许多基于模块度函数的蛋白质复合物挖掘算法[11]。Becker 等人[12]提出基于层次聚类的OCG(Overlapping cluster generator)算法,该算法根据Newman 的模块度函数将初始的重叠类迭代地融合到层次结构中,进而实现蛋白质聚类过程,然而该算法对复合物的模块规模较敏感,难以识别规模较小的复合物。郭茂祖等人[13]首先将识别稠密子图作为初始模块,然后根据模块度函数来合并初始模块,提出了复合物识别算法BMM(Based on protein complexes modularity function for merging modules)。Shen 等人[14]在引入外部紧密关联度和内部模块紧密关联度的基础上,设计了一种基于自适应密度模块化的蛋白质功能模块检测算法ADM(Adaptive density modularity)。在这个算法中,蛋白质可以自适应地选择留在当前的模块还是转移到另一个模块中。Shen 等人[15]设计了一种基于最佳邻节点和全局量化的复合物检测算法BN-LGQ(Best neighbour and local-global quantification)。该算法通过搜索当前簇的最佳邻节点和计算模块函数增量,进而确定是否将最佳邻节点加入簇中来扩展复合物。虽然基于模块度函数的蛋白质复合物挖掘算法已有了很大的进步,但是这些算法仅仅考虑了网络的拓扑特性,未考虑到PPI网络中富含的生物信息;且难以识别出重叠和规模较小的蛋白质复合物,算法的识别精度较低。虽然基于加权PPI网络的复合物识别取得了一定的成效,但是如何有效地构建加权PPI网络,如何降低复合物识别效果受假阳性和噪声数据的影响;如何解决基于模块度函数的蛋白复合物挖掘算法仅仅只考虑网络的拓扑特性而未考虑生物信息,以及难以识别出重叠和规模较小的复合物等导致的准确率、召回率不高以及执行效率低等缺陷,仍是亟待解决的问题。
针对以上问题,本文提出了一种基于模块度函数的加权蛋白质复合物识别算法IWPC-MF(Algorithm for identifying weighted protein complexes based on modularity function)。主要工作为:(1)融合点聚集系数改进边聚集系数,将改进后的边点聚集系数与基因共表达的皮尔逊相关系数相结合来构建加权蛋白质网络;(2)基于节点权重选取种子节点,遍历种子的邻居节点,设计节点间的相似度度量和蛋白质附着度来获取初始聚类模块;(3)设计基于紧密度的蛋白质复合物模块度函数来合并初始模块,并最终完成复合物的识别。实验结果表明本文算法运行效率高,聚类结果的准确率以及召回率较高。
2 相关定义
定义1(加权蛋白质网络)[16]设G=( )V,E,P 代表加权PPI 网络,V=( v1,v2,…,vn)代表蛋白质节点E=(e1,)代表蛋白质之间的相互作用,P=(p( e1),p( e2),…,是每条边存在的概率权重。
定义2(边聚集系数)[17]设无向图G=(V ,E,P ),其中表示节点u 和v 所共有的邻居节点数,Nu和Nv分别代表节点u 和v 的包括自身的邻居节点集合。则对于无向图中(u,v)的边聚集系数(Edge Clustering Coefficient,ECC)定义为:

定义3(皮尔逊相关系数)[18]给定k 个不同时刻的基因表达数据样本,则两个蛋白质节点u 和v 之间的皮尔逊相关系数(Pearson Correlation Coefficient,PCC)计算方式如式(2)所示:
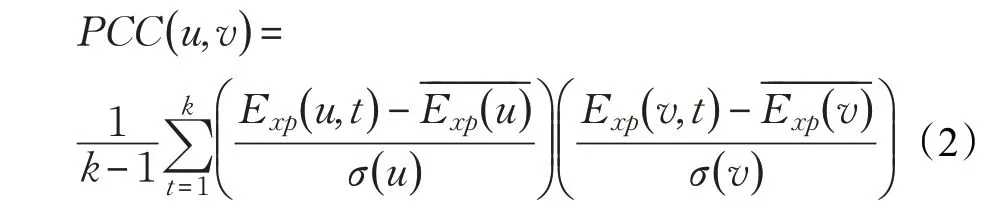
定义4(模块度函数)[19]给定PPI 网络G=(V ,E ),其中Aij表示节点i 和j 邻接关系,θ( ci,cj)表示节点i和j 的模块关系,di和dj分别代表节点i 和j 节点的度。则模块度函数(Modularity Function,MF)的定义如式(3)所示:

3 IWPC-MF算法
3.1 算法思想
针对蛋白质相互作用网络存在不稳定性,复合物的识别效果容易受到假阳性和噪声数据的影响;基于模块度函数的蛋白复合物挖掘算法只考虑网络的拓扑特性而未考虑生物信息以及难以识别出重叠和规模较小的复合物等问题,为提高算法的准确率、召回率、执行效率和降低假阳性的影响,本文提出了一种加权蛋白质复合物识别算法IWPC-MF。具体IWPC-MF算法思想为:首先以蛋白质相互作用网络为框架,融合点聚集系数改进边聚集系数得到边点聚集系数,利用边点聚集系数与基因共表达的皮尔逊相关系数衡量相互作用边的可靠性进而构建加权网络;其次利用节点权重选取种子节点,遍历种子节点的邻域节点,利用本文设计的节点间的相似度度量和蛋白质附着度来获取初始聚类模块;最后基于节点紧密度改进模块度函数,进而利用模块度函数将有重叠的初始模块进行合并,并最终完成复合物识别。
3.2 加权网络的构建
针对蛋白质相互作用网络存在不稳定性,复合物的识别效果容易受到假阳性假阴性和噪声数据的影响,本文基于PPI网络的拓扑特性,结合基因共表达的皮尔逊相关系数,对蛋白质相互作用网络进行加权,并且添加新的相互作用,能一定程度上增加蛋白质相互作用的可信度。
3.2.1 PPI网络的拓扑特性
由于蛋白质相互作用网络具有小世界特性和稀疏性,且存在一定比例的假阳性和假阴性,若两个蛋白质都同时第三个蛋白质发生相互作用,则这两个蛋白质间相互作用假阳性的可能性比较小,共同参与模块执行相同功能的概率比较大,因此,一对蛋白质之间相互作用的概率可以通过它们共有的邻居节点数量确定[20]。基于节点间共有的邻居数量的边聚集系数是PPI 网络中重要的拓扑特性,可以用于衡量测度图中的边聚集在一起的程度,还可以评估节点邻居之间的紧密程度。但是边聚集系数仅仅考虑边的重要性,没有考虑节点的重要性。因此,引入能够反映节点聚集程度的点聚集系数[21]对边聚集系数加以改进,提出一种融合节点和边的双重拓扑特性的边点聚集系数,则边点聚集系数度量如式(4)所示:

式(4)中,| Nu∩Nv|表示集合Nu除去和集合Nv的交集部分所剩余的蛋白质个数,| Nv-Nu|表示集合Nv除去和集合Nu的交集部分所剩余的蛋白质个数,Cu和Cv分别表示节点u 和v 的点聚集系数,其度量方式如式(5)所示:

式(5)中,i 可分别代表节点u 和v,| |Ni代表节点u 或v 的度,| |Ei表示由节点u 或v 的邻居节点之间组成的边的数目。
3.2.2 蛋白质的皮尔逊相关系数
由于高通量实验获得的蛋白质相互作用数据中存在着一定比例的假阳性和噪声数据,如果仅仅用网络的拓扑特性来衡量两个蛋白质之间的相互作用程度是比较片面的,因此本文引入定义3的基因共表达皮尔逊相关系数来衡量两个蛋白质之间的共表达程度进而增强相互作用之间的可靠性。
3.2.3 蛋白质网络的加权
基于蛋白质相互作用网络的拓扑特性,整合皮尔逊相关系数对网络进行加权,该加权策略不仅考虑了边和点聚集的重要性,还考虑到相互作用的蛋白质之间的基因共表达程度,共同体现蛋白质相互作用的可信度,其边加权的权重越大,可信度就越高。则对于PPI网络中任意两个蛋白质之间的相互作用(u,v)的权重计算如式(6)所示:

本文根据网络的拓扑特性和生物基因共表达皮尔逊相关系数构建加权网络,将边权值为0的边删除来降低噪声数据对挖掘蛋白质复合物检测结果造成的负面影响。
权值为0的边删除的必要性分析如下:
大量研究表明,通过高通量的生物实验方法获得的蛋白质相互作用数据中包含着较高比例的假阳性和假阴性,为了减少假阳性和假阴性造成的负面影响,本文通过综合考虑蛋白质网络的拓扑特性即蛋白质相互作用边之间的紧密联系以及节点的聚集系数来降低假阳性的影响,以及生物数据即皮尔逊相关数据来添加新的蛋白质相互作用,进而减少假阴性的影响。综合分析蛋白质相互作用的拓扑特性和生物特性来构建加权蛋白质网络,故将边权值为0的边作为假阳性而进行删除。
降低噪声数据对挖掘蛋白质复合物检测结果造成的负面影响等方面的作用分析:
为了进一步分析降低噪声数据对挖掘蛋白质复合物检测结果造成的负面影响,分别将边权重为0的边删除以及不删除边权重为0的边,利用本文改进的方法来挖掘蛋白质复合物,具体识别出的复合物的基本信息如表1所示。

表1 复合物的基本信息
从表1 可以看出,未删除边权重为0 的边的蛋白质复合物的平均蛋白质的个数为11.6,比删除边权重为0的边挖掘的复合物的平均个数要少,以及识别出的复合物比删除边权重为0 的复合物的个数要少。这是由于未删除边权重为0 的边中存在噪声数据实验结果容易受到假阳性的影响。故本文方法将边权重为0 的边删除可以有效地减少和控制噪声对于蛋白质复合物检测所产生的负面影响。
3.3 初始模块的形成
初始模块的形成过程主要包括节点种子的选取,根据节点间的相似性,遍历种子节点的邻域,计算蛋白质间的附着度进而形成初始模块。
基于蛋白质网络的拓扑特性和皮尔逊相关系数构建的加权网络,节点u 所有关联的边点聚集系数之和即节点权重定义如式(7)所示:

基于蛋白质复合物成簇出现且高度共表达,则蛋白质间的相似度度量如式(8)所示:

式(8)中,PCC( i,j )表示节点间的皮尔逊相关系数,PECC( i,j )表示节点i 和j 之间的结构相似性,Ni代表节点i 的邻居节点集合。PCC( i,j )和PECC( i,j )的差值表示节点共表达程度与拓扑结构相似性的差异,差异越小,说明这两个蛋白质无论是拓扑结构还是基因共表达信息的相似度都很接近,模块划分结构越加稳定。其值越大,说明这两个节点越有可能在同一模块内。
根据蛋白质节点之间的相似度度量,综合考虑节点与邻域节点间以及节点与邻域节点的邻接节点的相似度,设计出蛋白质附着度度量公式。
给定加权蛋白质子网络WG1=( )V1,E1,P1和节点i ∈V1,SM( )i,j 为节点间的相似性度量,加权网络G2=(V2,E2,P2)是包含G1中所有节点的邻居节点以及对应的相互作用边。则节点i 的的附着度计算如式(9)所示:

初始模块的形成思想如下:首先,基于蛋白质复合物是成簇出现且倾向于共表达的事实,利用式(6)来计算边的存在概率,构建加权网络;接着充分考虑节点之间的紧密程度以及共表达程度,对于加权网络中的任意一个节点,利用式(7)来计算该节点在其邻居图中的节点的权重,将加权网络中的边权值为0 和节点权重为0的作为噪声移除,同时按照WP( )u 值进行降序排列作为扩张的种子节点;最后遍历种子节点的邻域,根据相似度度量式(8)来计算节点间的相似性,同时根据式(9)计算蛋白质节点的附着度,将附着度大于阈值的节点加入到种子节点中,重复进行操作形成初始模块。
初始模块的形成过程形式化如下:
输入:蛋白质网络G( )
V,E ,附着度阈值δ,基因表达数据
输出:初始模块集合EM(1)构建加权蛋白质网络
①For each ( u,v )∈E do
② Compute PECC( u,v )by Eq(.4)
③ Compute PCC( u,v )by Eq(.2)④ Compute P( u,v )by Eq(.6)
⑤ If P( u,v )=0 do
⑥ Remove()//除去权值为0的相互作用边
⑦ End if
⑧End for
(2)选取种子节点
①L=∅ //种子节点集合
②For each vi∈V do
③ Compute WP( vi)by Eq(.7)
④ Sort()//将蛋白质节点按照WP( vi)值非递减排序WP( v1)≥WP( v2)≥…≥WP( vk)进入种子节点集合L 中。
⑤End for
(3)初始模块形成
①EM=∅,L={v1,v2,…,vk}
②For each vi∈L do
③G1={vi|dis( vi,va=1) }∪{vi}
④ If DS( vi,G1)>δ do
⑤ EM=EM ∪{vi}
⑥ End if
⑦End for
⑧ Return EM //输出蛋白质初始模块
3.4 初始模块的合并
大量研究发现,不同的蛋白质复合物之间可能存在重叠;大多数蛋白质复合物所包含的蛋白质节点数目较少;而且蛋白质复合物往往是倾向于成簇和高度共表达出现[13]。然而基于模块度函数的蛋白复合物挖掘算法存在仅仅只考虑网络的拓扑特性而未考虑生物信息以及难以识别出重叠和规模较小的复合物等问题,为提高算法的准确率和召回率,基于蛋白质网络的拓扑特性和基因共表达皮尔逊相关系数构建的加权网络,考虑到蛋白质复合物是节点紧密连接的稠密子图和蛋白质网络本身具有的小世界特性,提出基于紧密度[22]的蛋白质复合物合并模块度函数,若相邻的模块能够使模块度函数增加,则对模块进行合并进而得到蛋白质复合物。
紧密度是保证形成高内聚复合物的形成条件之一,则一个蛋白质节点v 到模块S 的紧密度F(v)的计算方式如式(10)所示:

基于紧密度的模块度函数计算公式如式(11)所示:

式(11)中,网络中边的数目为 ||E ,当前网络的模块数为| EM |,Si和Sj分别表示节点i 和j 所在的模块。
为了能够识别出更多规模较小和重叠的蛋白质复合物进而提高识别的精度,基于改进后的模块度函数,若初始两个模块合并能使模块度函数值增加,则合并这两个模块,并更新模块度函数值。
基于紧密度的模块度函数,具体模块合并过程形式化如下:
输入:加权蛋白质网络WG(V,E),初始模块集合EM
输出:蛋白质复合物集合C
(1)C=∅
(2)For i =1 to| EM |-1
(3)Compute DMF( EM′ )//计 算EMi和EMi+1合并后的EM′的模块度函数值
(4) If FMF( EM′ )>FMF( EM )do
(5) EMi=EMi∪EMi+1//删除EMi+1
(6) FMF( EM )=FMF( EM′)
(7) C=EM
(8) End if
(9)End for
(10)Return C //输出蛋白质复合物
3.5 IWPC-MF算法步骤
IWPC-MF算法具体的实现步骤如下所示:
输入:蛋白质网络G(V,E,基因表达数据
输出:蛋白质复合物C
(1)初始化参数:附着度阈值δ
(2)蛋白质复合物的挖掘
①For each ( u,v )∈E do
② 调用加权网络形成过程,获得加权网络WG(V,E)
③ For each vi∈V do
④ 调用种子节点形成过程,获得种子节点集合L
⑤ For each vi∈L do
⑥ 调用初始模块形成过程,获得初始模块集合EM
⑦For i=1 to| EM |-1
⑧调用初始模块合并过程,获得蛋白质复合物集合C
⑨End for
⑩ End for
⑪ End for
⑫End for
⑬Return C //得到蛋白质复合物
3.6 算法的时间复杂度
IWPC-MF 算法的计算复杂度由以下几个步骤构成:假设PPI 网络中节点数目为n,依据边点聚集系数以及基因表达数据构建加权PPI 网路的时间复杂度为O(n2);基于节点权重来选取种子节点的时间复杂度为O(n2);遍历种子节点的邻域,通过计算蛋白质节点之间的相似性,采用蛋白质附着度来形成初始模块的时间复杂度为O(n2);假设初始的模块数为h,则模块合并的时间复杂度为O(h2)。因此,IWPC-MF算法的时间复杂度为O(n2+h2)。由于h <n,所以IWPC-MF 算法的时间复杂度近似为O(n2)。而在GENA 算法中,算法的时间复杂度主要取决于初始化以及优化集群的过程,即O(Bn3);在WCOACH 算法中,算法的时间复杂度主要取决于初始核的检测和添加附件形成蛋白质复合物的过程,即O(τn3);在IMHRC 算法中,算法的时间复杂度主要取决于主要蛋白质集群形成以及合并修复集群的过程,即O(pγβn2);在ICMDS 算法中,算法的时间复杂度主要取决于复合核的形成以及冗余过滤,即O(n3);在OCG 算法中,算法的时间复杂度主要取决于极大团的形成以及合并模块的过程,即O(hn2);在BMM算法中,算法的时间复杂度主要取决于初始模块的形成以及合并模块的过程,即O(n2+h2),虽然该算法的复杂度和本文算法的时间复杂度一样,但该算法的识别精度以及匹配率较低;在ADM算法中,算法的时间复杂度主要取决于外部和内部紧密关联度的计算以及合并模块的过程,即O(n3);在BN-LGQ算法中,算法的时间复杂度主要取决于模块化增量计算以及模块形成过程,即O(n+φn+)。上述提及的nc、T 、τ、γ、β、B、φ 和χ 分别表示最大的复合物的蛋白质数目、基因表达时刻数、邻域亲和力阈值、中心获取阈值、中心移除阈值、预测到的模块数目、复合物形成前的数量和合并过程中蛋白质复合物减少的数量。
4 实验结果以及分析
4.1 实验环境
IWPC-MF 算法实验的编程环境为Python3.5.2;操作系统为Windows10 家庭中文版;内存12 GB;处理器为Intel®CoreTMi5-4200H CPU @ 2.8 GHz。
4.2 实验数据集
为验证本文提出算法的有效性,选用蛋白质相互作用数据相对完整和可靠的酵母蛋白质相互作用网络数据作为实验数据。具体实验数据如下所示:
(1)酵母PPI网络数据来源于DIP数据库[23],去除重复以及自相互作用,该数据库包含4 995个蛋白质和21 554对相互作用。
(2)实验采用的时序基因表达数据为GSE3431[24],包含7 079个蛋白质和36个时刻下的基因表达值。
(3)本文采用CYC2008[25]作为标准数据集,该数据集包含408个通过生物实验预测得到的蛋白质复合物。
4.3 评价指标
4.3.1 精度、召回率和F-measure 度量
为了评价本文算法的有效性,采用文献[26]的基于邻域亲和评分的精度(Precision)、召回率(Recall)和F度量(F-measure)指标来评价算法性能。邻域亲和评分是用来衡量预测的复合物与实际复合物的匹配度即重叠率,当重叠率OS(p,b)≥ω,输出最终的复合物,否则将该复合物删除,其定义为:


综合考虑精度和召回率对聚类结果的影响,采用F-measure 综合评估整体算法的性能。其计算公式如下:

4.3.2P-value值度量
随着蛋白质组学研究的深入,使得一个蛋白质与其功能注释相对应成为可能,蛋白质簇发生对于一个给定功能注释在统计学上的意义就可以通过一个超几何分布的等式来进行计算[28]:

其中,V 代表PPI 网络中包含的蛋白质总数,C 为预测挖掘出的复合物数目,F 为一个功能组数量,k 为C 中包含F 中的蛋白质数目。
4.4 参数选择
IWPC-MF 算法中,由于参数δ 的取值影响实验的聚类效果,因此本文在不同的δ 参数取值上独立运行20次实验,取20次实验的平均值进行分析。在图1中展示了δ 在不同取值下的F-measure 值和被识别匹配的蛋白质复合物比例变化情况,具体的实验结果如图1所示。

图1 F-measure 值和匹配的蛋白质复合物比例变化图
图1 可知,随着δ 从0 到0.2 逐渐增大,F-measure的值在δ 不同取值之下也逐渐增大,且F-measure 达到最大值0.482 9,实验识别的复合物和已知的复合物的匹配比例也逐渐增加,且达到最大值65.51%;随着δ 从0.2到1逐渐增大,F-measure 的值在δ 不同取值之下逐渐降低,实验识别出的复合物和已知的复合物的匹配比例也逐渐降低。这是因为本文融合边点聚集系数与基因表达数据构建加权网络,同时利用节点权重来选取种子节点,遍历种子节点的邻居时,计算节点之间的相似度,根据蛋白质附着度来形成初始模块,充分考虑了内部节点与外部节点之间的联系即全局一致性。随着附着度阈值的增大,算法识别的聚类数目逐渐增加,每个复合物中包含的蛋白质数目越少,相对应复合物的个数就会增加,但是当阈值增大到一定值时,扩展的种子邻域节点和种子节点之间的相互作用要求提高,导致挖掘复合物的精度逐渐增加,对挖掘出的蛋白质复合物会更严格,导致算法F-measure 值和匹配比例先增加后降低。通过观察发现存在一对合理取值即δ=0.2,使F-measure达到最大值0.482 9且匹配比例达到65.51%。故本文设置δ=0.2。
4.5 边点聚集系数度量的有效性分析
为了验证IWPC-MF 算法使用边点聚集系数PECC度量公式的有效性,分别基于使用边点聚集系数度量和皮尔逊相关系数IWPC-MF-PECC-PCC加权的IWPC-MF算法和未使用边点聚集系数即使用边聚集系数ECC和皮尔逊相关系数IWPC-MF-ECC-PCC加权的IWPC-MF算法,比较在算法识别的复合物与已知的复合物的匹配重叠率不同阈值下得到的F-measure 值和匹配比例,具体的实验结果如图2所示。

图2 不同加权策略挖掘的复合物对比结果
由图2 显示,使用边点聚集系数度量的IWPC-MF算法的F-measure 取值和匹配的蛋白质复合物比例都比未使用边点聚集系数的取值要高。在本文取值重叠率阈值为0.2 时,F-measure 的取值比未使用边点聚集系数提高3.62%,匹配的蛋白质复合物比未使用边点聚集系数度量加权提高4.65%。实验结果说明,使用改进的边点聚集系数的算法的聚类效果得到了提高。这是因为:基于复合物内部节点之间的紧密联系,IWPC-M算法在充分考虑网络的拓扑特性以及基因共表达程度时,还考虑到节点的聚集程度对复合物挖掘的影响,利用边点聚集系数和皮尔逊相关系数对蛋白质网络进行加权,进而利用种子节点实现复合物的挖掘。也进一步证明利用种子蛋白质能很好扩展为一个复合物。
为进一步检验边点聚集系数的有效性,分别使用不同加权方法与本文的IWPC-MF-PECC-PCC加权方法进行对比实验,图3是采用不同加权方法检测到蛋白质复合物与标准数据库的对比结果。从图3可以看出,使用IWPC-MF-ECC 加权的方法未识别出YHR081W 和YOL142W 两个蛋白质,是因为边聚集系数只考虑节点间边的紧密度,对网络的拓扑性分析比较单一;使用IWPC-MF-PECC 加权的方法未识别出一个蛋白质,这是因为边点聚集系数不仅考虑了节点间边的紧密关系,还考虑到了每个节点的重要性,对蛋白质网络的拓扑特性考虑得较全面,但忽略了蛋白质之间的生物特性;使用IWPC-MF-PECC-PCC加权的方法既考虑到网络的拓扑特性,同时又结合基因共表达的皮尔逊相关系数,对蛋白质网络的分析比较全面,能够更加贴近真实网络,因此最终的聚类效果较好。
4.6 加权蛋白质网络的性能分析
为了进一步分析IWPC-MF算法结合边点聚集系数和皮尔逊相关系数构建加权网络的性能,分别基于边点聚集系数和皮尔逊相关系数IWPC-MF-PECC-PCC加权的蛋白质网络、边点聚集系数IWPC-MF-PECC 构建的加权网络和皮尔逊相关系数IWPC-MF-PCC 构建的加权网络来挖掘蛋白质复合物,比较算法识别的复合物与已知的复合物在不同匹配重叠率阈值之下的匹配比例,具体的实验结果如图4所示。
由图4显示,本文使用边点聚集系数和皮尔逊相关系数IWPC-MF-PECC-PCC构建加权蛋白质网络挖掘复合物的检测效果明显优于其他加权策略,尤其在匹配重叠率阈值为0.2时,IWPC-MF算法使用IWPC-MF-PECCPCC加权识别的蛋白质复合物匹配比例分别比使用PECC加权和PCC加权识别的匹配比例高3.00%和5.83%。实验结果说明,结合边点聚集系数和皮尔逊相关系数的算法的聚类效果得到了提高。这是因为:基于边点聚集系数加权的蛋白质网络仅仅整合了边聚集系数和点聚集系数,因此只能反映PPI 网络的拓扑特性,考虑得比较单一;基于皮尔逊相关系数加权的网络仅仅只考虑到基因共表达程度,没有考虑网络的拓扑特性,实验结果存在较高的假阳性和假阴性数据;而基于复合物内部节点之间的紧密联系,IWPC-MF 算法在充分考虑网络的拓扑特性以及基因共表达程度时,还考虑到节点的聚集程度对复合物挖掘的影响,结合边点聚集系数和皮尔逊相关系数对蛋白质网络进行加权,有效地减少假阴性和假阳性带来的负面影响,因此本文的加权蛋白质网络的性能较好。

图3 不同加权方法识别nuclear exosome complex复合物

图4 不同加权网络策略的蛋白质匹配比例值的对比分析
为了减少假阳性和假阴性造成的负面影响,本文通过综合考虑蛋白质网络的拓扑特性即蛋白质相互作用边之间的紧密联系以及节点的聚集系数来降低假阳性的影响,以及生物数据即皮尔逊相关数据来添加新的蛋白质相互作用,进而减少假阴性的影响。综合分析蛋白质相互作用的拓扑特性和生物特性来构建加权蛋白质网络。为了进一步分析本文方法PPI 网络任意两个蛋白质之间的相互作用权重计算方式设计的准确性,采用本文的加权方式以及采用边点聚集系数构建加权网络来挖掘蛋白质复合物,具体检测结果如图5所示。通过具体实例分析DNA-directed RNA polymerase II complex复合物中任意边的相互作用,发现本文加权识别的复合物更加贴近真实网络。本文设计的加权方法更加准确。
4.7 优化的模块度函数的有效性分析
为了验证IWPC-MF算法使用优化的模块度函数FMF的有效性,分别基于优化的模块度函数FMF 的IWPCMF算法和未使用该优化函数即使用MF函数的IWPCMF算法,在DIP数据库独立执行20次进行复合物的识别,实验检测结果对比分析如图6所示。
图6 显示的是使用优化的模块度函数FMF 的IWPC-MF算法在precision、recall、F-measure 取值和匹配的蛋白质复合物比例与未使用该优化函数即使用MF函数的对比情况,其中使用FMF的precision 的取值比使用MF函数提高6.73%,recall 的取值比使用MF函数提高7.80%,F-measure 的取值比使用MF 函数提高7.40%,匹配的蛋白质复合物比使用MF函数提高6.73%。这是因为,本文根据使用FMF 函数来对初始模块进行合并,充分考虑网络的拓扑特性以及基因表达程度的同时,也考虑到复合物的重叠性以及复合物规模较小的性质,使得挖掘的复合物较准确,避免存在较高比例的复合物之间功能相似的模块,实验结果说明,使用FMF函数的算法的聚类效果较优。

图5 蛋白质边权重的准确性对比分析
在PPI 网络中,通过对蛋白质复合物的结构分析,可以发现多数蛋白质复合物的规模较小[13];而且同一个蛋白质可能属于不同功能的复合物,这些蛋白质往往具有多个生物功能,即蛋白质复合物之间可能会存在重叠。

图6 优化的模块度函数的对比分析
在标准408个复合物中,根据文献[13]的记录,标准复合物实际体积大于20 的只有不到10 个,半数以上的蛋白质复合物体积不大于3。为了验证本文识别蛋白质复合物的改进方法对规模较小复合物的识别能力,采用复合物体积来表示复合物中包含的蛋白质的个数,具体的蛋白质复合物体积分布直方图如图7所示。

图7 蛋白质复合物体积分布直方图
从图7可以看出,利用本文改进的方法检测到的体积大于21的不到9个,但是半数以上的复合物的体积不大于6。这说明多数蛋白质复合物的规模较小,且本文改进的方法可以识别出规模较小的复合物。

图8 蛋白质复合物体积分布直方图
从图8可以看出蛋白质复合物的重叠分布情况,其中重叠的蛋白质个数为2个和3个的蛋白质复合物数目最多,分别达到了124个和121,分别占本文识别的374个复合物的33.16%和32.35%。从图7和图8可知,本文改进的方法对规模较小和重叠蛋白质复合物的识别能力较优。这是因为本文综合考虑网络的拓扑特新和生物特性,以及使用基于紧密度的改进的模块度函数来合并初始模块,能够识别出重叠和规模较小的复合物。
4.8 算法性能的比较分析
本节将IWPC-MF 算法分别从精度、召回率和F-measure 的比较分析、聚类效果的比较分析和功能富集的比较分析与GENA[5]、WCOACH[6]、IMHRC[7]、ICMDS[9]、OCG[12]、BMM[13]、ADM[14]、BN-LGQ[15]算法进行比较分析。重复迭代次数20次。实验使用到的参数设置δ=0.2。
(1)精度、召回率和F-measure 的比较分析
为了验证本文算法的性能,将IWPC-MF 算法与其他8种算法在DIP数据上独立运行20次,取实验结果的平均值进行分析,得到各个算法识别的复合物基本信息以及实验评价指标对比分析如表2和图9所示。

表2 各算法识别的复合物的基本信息

图9 算法性能比较关系图
在表2 中,PM 表示算法识别出的复合物总数,average 是指每个簇中的蛋白质平均个数。由表2可以知道,IWPC-MF 算法共识别374 个复合物,每个复合物平均包含13.5 个蛋白质,其中245 个预测结果较准确,标准集合中的156个复合物可以被算法准确识别到,是所有算法中识别匹配最多的。这是因为IWPC-MF算法在构建加权蛋白质网络的时候不仅考虑了蛋白质网络本身的拓扑特性,还考虑到蛋白质之间的基于共表达程度,这样降低了假阳性和噪声数据对实验结果产生的负面影响;同时利用优化后的模块度函数合并初始模块时,充分考虑蛋白质复合物之间的重叠性以及大多数复合物规模较小的特性,这样可以提高识别的准确性。因此,本文提出的IWPC-MF算法的挖掘效果较好。
图9显示了各种算法在DIP数据集中识别的复合物的结果。从图中可以清晰地发现IWPC-MF 算法在精度、召回率和F 度量指标上取得较好的结果。具体来说,IWPC-MF 算法的精度为65.51%,相比较GENA、WCOACH、IMHRC、ICMDS、OCG、BMM、ADM 和BNLGQ 算法分别提高了39.69%、57.75%、14.17%、3.74%、43.93%、40.44%、44.66%和40.67%;召回率为38.24%,相比较GENA、WCOACH、IMHRC、ICMDS、OCG、BMM、ADM 和BN-LGQ 算法分别提高了79.31%、90.24%、52.94%、8.33%、65.96%、59.18%、83.53%和54.46%;F 值度量为48.29%,相比较GENA、WCOACH、IMHRC、ICMDS、OCG、BMM、ADM 和BN-LGQ 算法分别提高了64.71%、78.27%、38.65%、6.64%、57.84%、52.27%、69.21%和49.38%。实验结果表明,使用本文算法挖掘蛋白质复合物的聚类精度、召回率和F-measure 相比较其他8种算法都得到了提高。这是因为,在GENA算法中,使用贪婪方法初始化集群,在聚类系数的基础上选取种子节点,仅仅考虑了网络的拓扑特性,挖掘的效果存在大量的重叠模块;在WCOACH 算法中,仅仅利用GO 信息来构建加权网络,缺乏考虑蛋白质网络本身的拓扑特性以及特征,且在聚类的时候,若选取的核心节点较为相似,则会挖掘出大量重叠的模块,最终导致挖掘的准确性降低;在IMHRC算法中,构建加权PPI网络时,仅仅考虑节点度即网络的拓扑结构,没有融合生物信息,考虑构建的加权PPI 网络比较单一,使得挖掘聚类效果不佳。在ICMDS 算法中,基于边聚集系数和皮尔逊相关系数,在计算相互作用的功能相似性时引入了自定义的参数,导致挖掘效果受参数的影响比较大;在OCG、BMM、ADM和BN-LGQ算法中,仅仅只考虑网络的拓扑特性而未考虑生物信息以及难以识别出重叠和规模较小的复合物。而本文是综合考虑网络的拓扑结构和生物基因表达信息,基于边点聚集系数和皮尔逊相关系数来构建加权网络;同时根据节点权重选择种子节点,遍历种子节点的邻域,利用蛋白附着度来形成初始模块;最后利用改进的模块度函数合并有重叠的模块,可以较为精确和快速地挖掘出蛋白质复合物。因此,本文提出的算法的聚类效果较好。
(2)聚类效果的比较分析
为了评估本文提出的IWPC-MF 算法的聚类效果,将本文算法与其他8种算法挖掘的Elongator holoenzyme复合物可视化,对比分析聚类效果,聚类聚类结果的对比如图10所示。

图10 各个算法的复合物挖掘可视化比较图
图10 显示了不同算法检测到的Elongator holoenzyme复合物结果,图(a)是该标准复合物所包含的蛋白质相互作用情况;图(b)是本文算法的检测结果;图(c)是GENA 算法的检测结果;图(d)是算法WCOACH 的检测结果;图(e)是IMHRC算法的检测结果;图(f)是算法ICMDS的检测结果;图(g)是OCG算法的检测结果;图(h)是BMM算法的检测结果;图(i)是ADM算法的检测结果;图(j)是BN-LGQ 算法的检测结果。通过图10显示,本文算法能够准确地识别蛋白质复合物;GENA算法识别出标准复合物中的6个蛋白质,但是也包含了2 个非Elongator holoenzyme 复合物内的蛋白质;算法WCOACH 识别出标准复合物中的5 个蛋白质;算法IMHRC识别出标准复合物中的6个蛋白质,但是也包含了3 个非Elongator holoenzyme 复合物内的蛋白质;ICMDS算法也正确识别出标准复合物中的6个蛋白质;OCG 算法识别出标准复合物,但是也包含了4 个非Elongator holoenzyme复合物内的蛋白质;BMM算法识别出标准复合物中的6 个蛋白质,但是也包含了5 个非Elongator holoenzyme复合物内的蛋白质;ADM算法识别出标准复合物中的6 个蛋白质,但是包含了4 个非Elongator holoenzyme 复合物内的蛋白质;BN-LGQ 算法识别出标准复合物中的6 个蛋白质,但是也包含了1个非Elongator holoenzyme复合物内的蛋白质。实验结果表明,本文算法挖掘的蛋白质复合物聚类效果较好。这是因为,本文通过蛋白质网络的拓扑特性和基因表达信息来构建加权网络,可以降低假阳性和噪声数据的负面影响;同时根据节点权重选择种子节点,遍历种子节点的邻域,利用蛋白质附着度来形成初始模块,综合考虑蛋白质复合物是稠密且内部紧密连接的特性,以及蛋白质复合物成簇出现和高度共表达的特性;将得到的初始模块利用改进的模块度函数进行合并,充分考虑到蛋白质复合物的重叠性以及规模较小的特性,同时也避免算法重复的挖掘过程。实验结果表明,本文算法在识别蛋白质复合物上具有较好的聚类效果。
(3)功能富集的比较分析
为了测试算法识别的复合物的生物学意义,采用复合物的低P-value 值的功能富集分析。 P-value 值越小表明该复合物具有很高的统计学意义。若一个模块的P-value <0.01,则认为这个复合物是显著的[29]。显著的复合物数量在识别出的复合物总数中所占的比例可以很好地评价各个算法的整体性。具体各个算法性能比较如表3所示。

表4 IWPC-MF算法识别的复合物实例

表3 各个算法识别的复合物的显著性统计信息
在表3中,PM 表示算法识别出的复合物总数,SC表示具有生物显著意义的蛋白质复合物数目。IWPCMF算法识别的复合物数目中显著性复合物的比例达到85.29%,相比较GENA、WCOACH、IMHRC、ICMDS、OCG、BMM、AD 和BN-LGQ 算法分别提高了83.22%、14.81%、73.42%、3.19%、82.24%、70.58%、77.28%和53.62%。由此可见,IWPC-MF 算法识别出的复合物具有很强的生物统计学意义。这是因为本文算法在构建加权网络的时候,综合考虑网络的拓扑特性和基因共表达程度,同时根据蛋白质附着度利用种子节点形成初始模块,最后综合考虑复合物的重叠性和规模较小的性质,利用基于紧密度的模块度函数实现复合物的挖掘。最终导致聚类效果较好,执行效率高,挖掘的生物蛋白质复合物更具有生物统计意义。
表4具体给出本文IWPC-MF算法识别出的复合物实例,其中OA 表示算法识别复合物的匹配率,OM 表示的是正确匹配的蛋白质个数,Predicted protein 表示组成复合物的所有蛋白质,加粗部分表示被匹配的蛋白质。从表4 可以看出,当P-value=2.22E-18 时,本文算法识别的NatC 复合物的匹配率达到了0.82,正确匹配的蛋白质个数是9,这是因为YGR134W和YNL288W蛋白质与复合物内部连接比较松散。由此可见,IWPCMF算法识别的蛋白质复合物效果更好。
5 结束语
本文在结合边点聚集系数与基因表达数据构建的加权蛋白质网络基础上,提出一种基于模块度函数的加权蛋白质复合物识别算法IWPC-MF。基于节点权重选取种子节点,遍历种子的邻居节点,设计节点间的相似度度量和蛋白质附着度来获取初始聚类模块;设计基于紧密度的蛋白质复合物模块度函数来合并初始模块,并最终完成复合物的识别。为了评估算法的性能,本文将IWPC-MF算法与其他8种算法进行了对比,实验结果表明,IWPC-MF 算法具有更高的准确率、召回率,识别的复合物具有更强的生物统计意义。今后可以将IWPC-MF算法应用于疾病预测和关键蛋白质识别等相关研究中。
