直线型铂炔化合物的合成及其成凝胶性能研究
2020-01-06范建强曾旺徐潇杨凌辉余威张灯青
范建强,曾旺,徐潇,杨凌辉,余威,张灯青
(东华大学化学化工与生物工程学院,上海 201600)
铂炔配合物是利用金属离子和配体的配位而形成的一类化合物,金属与配体的配位键键能介于共价键键能与其他非共价键键能之间[1],而且配位键具有明确的方向性[2]。Yam[3-4]、Stang[5-8]、Fujita[9-14]等课题组报道了各种各样的金属铂配合物,使得金属有机材料得到了快速的发展。金属配位化合物能够表现出各种各样的性能,如光学性能、催化性能、氧化还原性能等[15]。Yam报道的许多金属铂配位有机物能够在有机溶剂中形成稳定的热可逆盐,形成Pt…Pt作用和π-π相互作用,在一些相变过程中显现出明显的颜色变化,这一性质可被用作微环境变化的有效报告物。金属配合物当中的金属凝胶配位化合物在近些年被许多科研工作者们所关注,Kawano等[16]在银离子和氮原子配位形成凝胶稳定性关系方面做了开创性的工作。此外,还有中科院长春应化所高连勋等[17]报道了金属有机凝胶做成纳米纤维,北京大学唐黎明教授等[18]利用金属有机凝胶做成高聚物中的单体封端剂等。金属配合物的合成是这些研究的基础,因此金属配合物在构筑新颖的金属有机材料方面具有重要的意义。
如图1所示,本文利用配位键定向键合的配位方式形成直线型铂炔化合物,通过酰胺基团引入氢键形成金属凝胶化合物1d,并用扫描电镜观察了其在固态下的微观结构。实验中所需要的原料a通过文献中的合成方法[19]来制备。
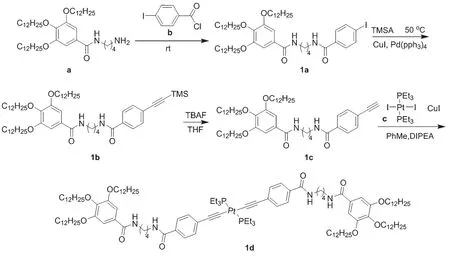
图1 化合物1d的合成
1 材料与方法
1.1 仪器与试剂
Bruker-400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标),HR-MS(ESI)Thermo Scientific Q Exactive HF Orbitrap-FTMS,HITACHI-S-4800型FE-SEM,电子分析天平(上海力衡仪器仪表有限公司)。
对碘苯甲酰氯(b)、对溴苯甲酰氯(d)、四三苯基膦钯、十二烷基溴、没食子酸甲酯、氢氧化钾、氟化四丁基铵、三乙胺、四氢呋喃、甲苯、二氯甲烷、导电胶以及二异丙基胺均购于上海泰坦科技有限公司;三甲基硅基乙炔、碘化亚铜购于百灵威科技有限公司。实验试剂和药品均为分析纯。
1.2 合成工艺
1.2.1 化合物1a的合成
向干燥的两口圆底烧瓶中加入357 mg(1.34 mmol)对碘苯甲酰氯和1 000 mg(1.34 mmol)化合物a,氮气保护下注入新蒸的二氯甲烷(10 mL)和三乙胺(2 mL)。室温下搅拌12 h后减压蒸除反应液,用二氯甲烷萃取(3×15 mL),合并有机相,用饱和食盐水洗涤,无水Na2SO4干燥,蒸除溶剂,残余物经硅胶柱层析[洗脱剂体积比V(EA)∶V(PE)=1∶4]纯化得到白色固体1a,产率为80%。通过400 MHz的核磁共振1H NMR(400 MHz, CDCl3为溶剂),δ(化学位移值):7.97 (d, J=1.1 Hz, 2H), 7.77 (s, 1H),7.10 (d, J=5.0 Hz, 1H), 7.02 (s, 2H), 6.53 (d, J=5.8 Hz,1H), 4.00 (dd, J=14.5, 6.7 Hz, 6H), 3.52 (dd, J=12.3,5.9 Hz, 4H), 1.85-1.74 (m, 6H), 1.72 (s, 4H), 1.44 (dd,J=14.0, 6.2 Hz, 6H), 1.27 (d, J=9.6 Hz, 48H), 0.87 (d,J=7.0 Hz, 9H);通过400 MHz的核磁共振,13C NMR(101 MHz, CDCl3为溶剂) δ(化学位移值): 167.68,166.98, 153.10, 141.15, 137.71, 133.93, 129.32, 128.70,105.75, 98.32, 73.52, 69.35, 49.26, 39.63, 33.85, 31.94,30.34, 29.72, 29.70, 29.67, 29.66, 29.60, 29.44, 29.42,29.39, 29.38, 26.89, 26.11, 25.58, 24.90, 22.70, 14.11;通过电喷雾高分辨质谱HR-MS (ESI) m/z: 理论值:975.6034, 实测值:975.6033。
1.2.2 化合物1b的合成
把化合物1a 800 mg(0.821 mmol)、CuI 23.4 mg(0.123 mmol)、Pd(pph3)4142 mg(0.123 mmol)加入到干燥的两口圆底烧瓶中,置换气3次后用一次性注射器注入新蒸的四氢呋喃(4 mL)和三乙胺(6 mL),常温下搅拌0.5 h后再注入三甲基硅基乙炔(97.0 mg),然后在50 ℃条件下反应12 h,反应结束后蒸除反应液,用二氯甲烷萃取(3×15 mL),合并有机相,用饱和食盐水洗涤,经无水Na2SO4干燥,蒸除溶剂,残余物经硅胶柱层析[洗脱剂体积比V(DCM)∶V(MeOH)=50∶1]纯化得到化合物1b为白色固体,产率75%。通过400 MHz的核磁共振1H NMR (400 MHz, CDCl3为溶剂) δ(化学位移值): 7.75 (d, J=8.3 Hz, 2H), 7.51 (d, J=8.2 Hz,2H), 7.03 (s, 2H), 6.63 (t, J=5.7 Hz, 2H), 4.03-3.97(m, 6H), 3.55-3.50 (m, 4H), 1.82-1.76 (m, 6H), 1.72(s, 4H), 1.46 (d, J=7.2 Hz, 6H), 1.26 (s, 48H), 0.87 (d,J=4.8 Hz, 9H), 0.26 (s, 9H); 通过400 MHz的核磁共振13C NMR (101 MHz, CDCl3) δ(化学位移值):167.75, 167.16, 153.24, 141.28 , 134.15, 132.21, 129.51,126.98, 126.55, 105.89, 104.31, 104.00, 97.11, 73.64,69.50, 33.96, 32.08, 30.48, 29.90, 29.89, 29.88, 29.87,29.85, 29.81, 29.80, 29.74, 29.57, 29.55, 29.54, 29.52,27.23, 26.94, 26.25, 25.70, 25.02, 22.84, 14.25; 通过电喷雾高分辨质谱HR-MS (ESI) 理论值:945.7455, 实测值:945.7454。
1.2.3 化合物1c的合成
称取化合物1b 500 mg(0.529 mmol),加入干燥的两口圆底烧瓶中,置换气3次后用一次性注射器注入新蒸的四氢呋喃(5 mL),氟化四丁基胺166 mg(0.634 mmol)溶于四氢呋喃中后滴加到反应瓶中,滴加结束后在室温下反应1.5~2.0 h。反应结束后蒸除反应液,用二氯甲烷萃取(3×15 mL),合并有机相,用饱和食盐水洗涤,经无水Na2SO4干燥,蒸除溶剂,残余物经硅胶柱层析[洗脱剂体积比V(DCM)∶V(MeOH)=50∶1]纯化得到化合物1c为白色固体,产率为82%。通过400 MHz的核磁共振1H NMR (400 MHz, CDCl3为溶剂) δ(化学位移值): 7.79 (d, J=7.8 Hz, 2H), 7.48 (d, J=7.8 Hz, 2H), 7.27 (s, 1H), 7.21 (s,1H), 7.07 (s, 2H), 3.95 (dd, J=13.4, 6.7 Hz, 6H), 3.44 (s,4H), 3.18 (s, 1H), 1.76-1.71 (m, 6H), 1.64 (s, 4H), 1.41(s, 6H), 1.26 (s, 48H), 0.88 (t, J=5.9 Hz, 9H)。
1.2.4 化合物1d的合成
称取化合物1c 330 mg(0.378 mmol)、化合物c 129 mg(0.189 mmol)、CuI 7.20 mg(0.038 mmol)一并加入干燥的两口圆底烧瓶中,置换气3次后用一次性注射器注入干燥过的甲苯(6 mL)和二异丙基胺(4 mL)。在30 ℃条件下反应3 d,反应结束后蒸除反应液,用二氯甲烷萃取(3×15 mL),合并有机相,用饱和食盐水洗涤,经无水Na2SO4干燥,蒸除溶剂,残余物经硅胶柱层析[洗脱剂体积比V(DCM)∶E(MeOH)=50∶1]纯化得到化合物1d为淡黄色固体,产率65%。通过400 MHz的核磁共振1H NMR (400 MHz, CDCl3为溶剂) δ(化学位移值): 7.66 (d, J=8.4 Hz, 4H), 7.30 (d, J=8.2 Hz, 4H), 7.06 (s, 4H), 6.84 (t,J=5.6 Hz, 2H), 6.56 (s, 2H), 4.01-3.97 (m, 12H), 3.49(d, J=5.1 Hz, 8H), 2.17 (ddt, J=11.1, 7.4, 3.7 Hz, 12H),1.81-1.74 (m, 12H), 1.69 (s, 8H), 1.47-1.42 (m, 12H),1.26 (s, 96H), 1.23-1.18 (m, 18H), 0.87 (d, J=6.1 Hz, 18H).通过400 MHz的核磁共振13C NMR (101 MHz, CDCl3为溶剂) δ(化学位移值): 167.56, 153.07,141.05, 140.96, 130.81, 129.45, 126.71, 105.72, 73.49,69.30, 39.83, 39.55, 31.94, 30.35, 29.76, 29.75, 29.74,29.73, 29.71, 29.69, 29.68, 29.67, 29.61, 29.44, 29.40,29.39, 27.41, 26.65, 26.12, 22.70, 16.63, 16.46, 16.28,14.12, 8.36.31P NMR (162 MHz, CDCl3) δ (ppm): 11.21(s,1JPt-P=245 2.785); 通过电喷雾高分辨质谱 HR-MS(ESI) m/z: 理论值:217 4.5339, 实测值:217 4.5338。
1.3 1d成凝胶性能测试及其扫描电镜观察
用称量天平称取5 mg的化合物1d后加入到5 mL的白色样品瓶中,再加入1 mL的有机溶剂,盖上样品的瓶盖后加热溶解,然后自然冷却至室温,观察各个样品的溶解情况及其在室温下的状态。拍摄电镜之前的化合物1d先捣成粉末状,再将其转移到导电胶上,并均匀铺成薄薄的一层,导电胶置于样品台上,制备好的样品进行喷金处理,然后进行扫描电镜拍摄。
2 结果与讨论
2.1 反应原料和溶剂的优化
以化合物a和b的反应为研究对象,考察了原料和溶剂的种类对化合物1a产率的影响,结果见表1。从表1可以发现反应在几种溶剂中均能进行,但在二氯甲烷中的产率最高。在原料选择上可以看出其他条件相同时,以对碘苯甲酰氯为原料比对溴苯甲酰氯为原料的产率要高。因此,合成化合物1a的最佳反应条件是以二氯甲烷为溶剂,对碘苯甲酰氯为反应原料,反应温度为25 ℃,反应时间为12 h。

表1 溶剂和原料的优化
2.2 反应催化剂和溶剂比例的优化
以化合物1a和三甲基硅基乙炔反应为研究对象,考察了催化剂用量和溶剂比例对化合物1b产率的影响,结果见表2。从表2中可见当CuI和Pd(pph3)4的用量为15%时,化合物1 b的产率达最大值;在此基础上继续优化溶剂体积比,可以看出四氢呋喃和三乙胺体积比为2∶3时,化合物1b的产率最高。因此,该反应的最优条件是CuI和Pd(pph3)4用量为15%,四氢呋喃和三乙胺溶剂体积比是2∶3,反应温度为50 ℃,反应时间为12 h。

表2 催化剂和溶剂比例的优化
2.3 化合物1d成凝胶性能测试及其在扫描电镜下的微观结构
常温下,化合物1d在各种溶剂中的状态如表3所示。化合物1d只有在低极性的甲基环己烷溶液中能够形成凝胶(化合物1d的凝胶状态见图2),在其他溶剂中只产生沉淀或呈现溶液状态。如图3所示,在扫描电镜下,化合物1d在甲基环己烷溶液中形成的凝胶固体是堆积状的。在凝胶状态下,溶剂完全挥发干后,化合物1d呈现出膜状,所以没有拍摄到其微观结构。

图2 化合物1d在甲基环己烷中形成的凝胶

图3 化合物1d固态下的SEM图
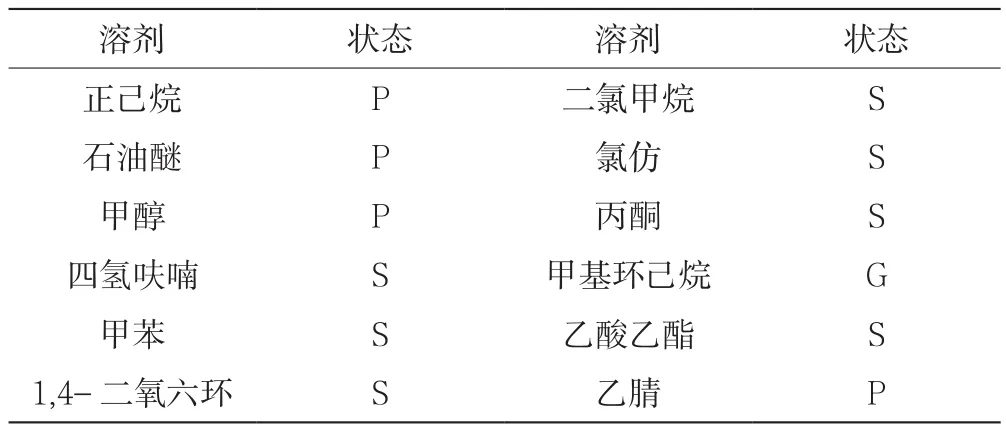
表3 化合物1d在不同溶剂中成凝胶的能力
3 结论
通过对反应条件的优化,从原料a开始依次经过酰胺缩合、Sonogashira偶联、脱三甲硅基保护基以及Hagihara偶联反应,最终以65%的产率得到了最终产物1d。中间产物1c末端炔烃上氢的化学位移值在3.18处,产物1d的1H NMR谱图中3.18处没有氢信号峰,说明1c完全参与了反应。此外,产物1d的磷谱和碳谱结果也表明1d的纯度较好。因为本研究中所做的铂炔化合物1d能够在低极性有机溶剂中形成凝胶,所以其在研究溶液极性对分子聚集行为方面具有一定的科研价值和应用前景。
