乙肝病毒(HBV)感染对肝细胞癌(HCC)患者A-to-I RNA编辑的影响
2019-12-05马婷黄海郝沛
马 婷 黄 海 郝 沛
(1上海大学生命科学学院发育神经生物学实验室 上海 200444; 2中国科学院上海巴斯德研究所病原大数据实验室 上海 200031)
肝细胞癌(hepatocellular carcinoma,HCC)是原发性肝癌中最主要的组织学亚型[1]。纤维板层肝细胞癌(fibrolamellar-HCC,FL-HCC)是HCC的一种特殊类型,常见于青年群体,通常不伴随肝硬化和乙型肝炎病毒(hepatitis B virus,HBV)感染[2-3]。已知HBV感染后会增加HCC的发病率[4]。
RNA编辑是指通过转录后修饰引起转录RNA序列改变,致使RNA携带信息改变的生物学现象[5]。脊椎动物中最常见的RNA编辑类型是腺嘌呤(adenine,A)转变为次黄嘌呤(inosine,I),即A-to-I RNA编辑。A-to-I RNA编辑发生在具有双链特征的RNA区域。底物RNA在腺苷酸脱氨酶(adenosine deaminases acting on RNA,ADAR)催化下将A水解脱氨基转化为I,后续翻译过程中被识别为鸟嘌呤(guanine,G)[6]。在病毒感染期间,不同的病毒与宿主组合后,ADAR可能发挥促病毒或抗病毒作用[7-8]。目前对HCC患者RNA编辑的研究主要集中在癌组织和正常组织间的编辑活性变化[9-10],而HBV感染对HCC患者A-to-I RNA编辑事件的影响尚无研究报道。
我们从数据库下载了HBV阴性和阳性患者的转录组数据[11-12],采用SPRINT软件[13]鉴定A-to-I RNA编辑事件,比较正常组织和癌组织中HBV阴性、HBV阳性样本组的ADAR1表达值及共有RNA编辑位点(RNA editing site,RES)的编辑水平差异情况。本研究首次分析了HBV感染的HCC患者A-to-I RNA编辑的变化情况,对深入研究HBV感染对HCC发生发展的分子作用机制有一定的意义。
材料和方法
材料转录组数据来自美国国家生物技术信息中心(National Center of B-iotechnology Information,NCBI)的GEO数据库,数据集编码为GSE63018和GSE77509。为区分两套数据,分别用FL-HCC和HCC来表示。FL-HCC数据集为2*50的双端非特异性测序数据,测序数据来自FL-HCC患者,由于该类HCC的特殊性,8位患者均为HBV阴性。HCC数据集则为2*100的双端非特异性测序数据,包含19位HBV阳性患者和1位HBV阴性患者。每位患者均有配对的正常和癌症肝组织的测序数据。
A-to-I RESs的鉴定和注释采用SPRINT 0.7.16软件鉴定RNA编辑事件。将匹配到参考基因组的读段(reads)和未匹配、处理后二次匹配的读段分为两条检测途径进行位点寻找,因此该法能找出数目更多的位点用于后续分析。在SPRINT中使用Burrows-Wheeler algorithm (BWA)算法[14]进行比对。人类参考基因组和基因组注释信息版本为hg19。采用Annovar软件对得到的位点进行注释[15]。
基因差异分析使用Trim_galore v0.4.3(http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/)去除测序质量低于20的读段,得到清洁数据。采用“-fastqc”参数对清洁数据进行测序质量评估。采用Hisat2 v2.0.5[16]比对到人类参考基因组得到SAM文件。用Samtools v1.5[17]的“samtools view”和“samtools sort”命令将SAM文件转化为BAM文件并进行排序。使用Picard v1.127软件(https://broadinstitute.github.io/picard/)去除样本制备过程中产生的PCR重复序列。HTSeq-0.6.1[18]用于计算基因比对到每个基因的读段数。使用转录组表达定量值CPM(counts per million)来实现基因表达值的标准化,即原始读段数÷总读段数*1 000 000。
标准化编辑水平计算编辑水平定义为每个发生A-to-I RNA编辑的读段数与比对到该位点的所有读段数的比值。为更好比较不同患者的编辑水平变化,我们选出正常组织和癌组织共有的RES。将共有RES最高的值作为标准,其他RES与之相除,得到标准化编辑水平[19]。
GO富集分析使用R包clusterProfiler软件[20]对每个患者正常组织和癌症肝组织的RES所在基因分别进行GO富集分析。找出在HBV阴性和HBV阳性样本组中均出现的GO富集通路,其中校正后P<0.05的通路为显著富集通路。
图形展示和统计学方法使用R开源包ggplot2和GraphPad prism8.0.1绘制图片。本研究在R环境下进行统计学分析,相关性检验使用Kruskal-Wallis检验,P值阈值设置为0.1。
结 果
两套数据集A-to-I RESs概况我们检测了FL-HCC和HCC数据集中56个样本的RNA编辑事件,分别找出299 528和122 265 509个RES(表1)。为验证RES的可靠性,将每个样本的检测位点与目前主要的RES数据库DARNED(http://darned.ucc.ie/)、RADAR(http://rnaedit.com/)、REDIportal(http://srv00.recas.ba.infn.it/py_script/REDIdb/)以及3个数据库的并集进行比较(表1)。结果发现,FL-HCC数据集与数据库的平均交集为76.04%,而HCC数据集与数据库的平均交集仅为52.47%。这是由于数据库中的位点主要是匹配到参考基因组的读段所找出的RES,而SPRINT还包含未匹配到基因组而进行二次匹配的读段所找出的RESs。所以两套数据集与数据库的交集并不高,这说明相对于以往的RES鉴定方法[21-22],本研究可检测出更多的RES来进行后续分析。
为进一步验证检测位点的准确性,我们分析了两套数据集RES的位点分类特征(图1)。将RES分为Alu区、重复非Alu (repetitive non Alu,REP-NON-Alu)区和非重复(non repetitive,NON-REP)区,FL-HCC和HCC数据集分别有超过72%和82%的位点位于Alu区,两套数据集RES的分布特征具有一致性。这说明由于Alu区的序列具有相似特征,其在基因和基因间易形成双链RNA,是ADAR酶的理想底物。因此,大部分位点发生在基因组的Alu区域,该现象与已有研究结果一致[23]。大部分位点分布在内含子和基因间区域,其次是3’-UTR和非编码RNA(non coding RNA,ncRNA)区域,外显子和5’-UTR区域分布最少。这些分布特性都表明我们所鉴定出的位点具有可信度。
FL-HCC:RNA-seq data collected from GSE63018;HCC:RNA-seq data collected from GSE77509.SPRINT:Number of RESs identified by SPRINT.
ADAR酶表达水平的变化HCC组织中ADAR1表达量升高与RNA编辑水平上调有明显相关性[9],但HBV感染后HCC样本ADAR1的表达变化与RNA编辑水平之间的关系尚不清楚。我们将两套数据集的样本按照不同组织来源和HBV感染情况,分为HBV阴性正常组织(HBV-N)、HBV阴性癌组织(HBV-T)、HBV阳性正常组织(HBV+N)和HBV阳性癌组织(HBV+T)等4个样本群体。考虑到两套数据集的批次效应,我们使用标准化的基因表达值来观察在不同组织和HBV感染情况下ADAR1的表达变化。在比较HBV-N和HBV+N、HBV-T和HBV+T群体后,我们发现在正常和癌组织中HBV感染均引起ADAR1的表达上调。在HBV阴性群体中,ADAR1在正常组织和癌组织之间无明显差异。在HBV阳性群体中,RNA编辑酶的整体活性更高,且癌组织的ADAR1表达水平相对于正常组织有明显上升(图2A)。这都说明HBV感染促进ADAR1表达,该现象在癌组织中更显著。此外,我们还观察到不同样本群体中ADAR2和ADAR1的表达趋势并不一致(图2B)。在HBV阴性群体中ADAR2在癌组织中的表达水平相对于正常组织更高,而HBV阳性群体中则相反。在不同组织中,HBV感染后ADAR2的表达水平更低。总体上,ADAR2的表达值明显低于ADAR1的表达值,这表明在催化编辑反应中ADAR1发挥主要作用,ADAR2的作用有待进一步分析研究。
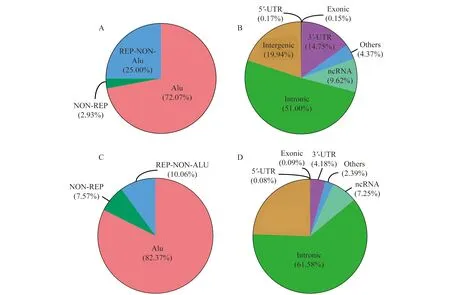
Distribution (A) and genomic distribution (B) of RESs in FL-HCC data set;distribution (C) and genomic distribution (D) of RESs in HCC data set.
图1 FL-HCC和HCC数据集中A-to-I RESs分布情况
Fig 1 The distribution of A-to-I RESs in FL-HCC and HCC data sets
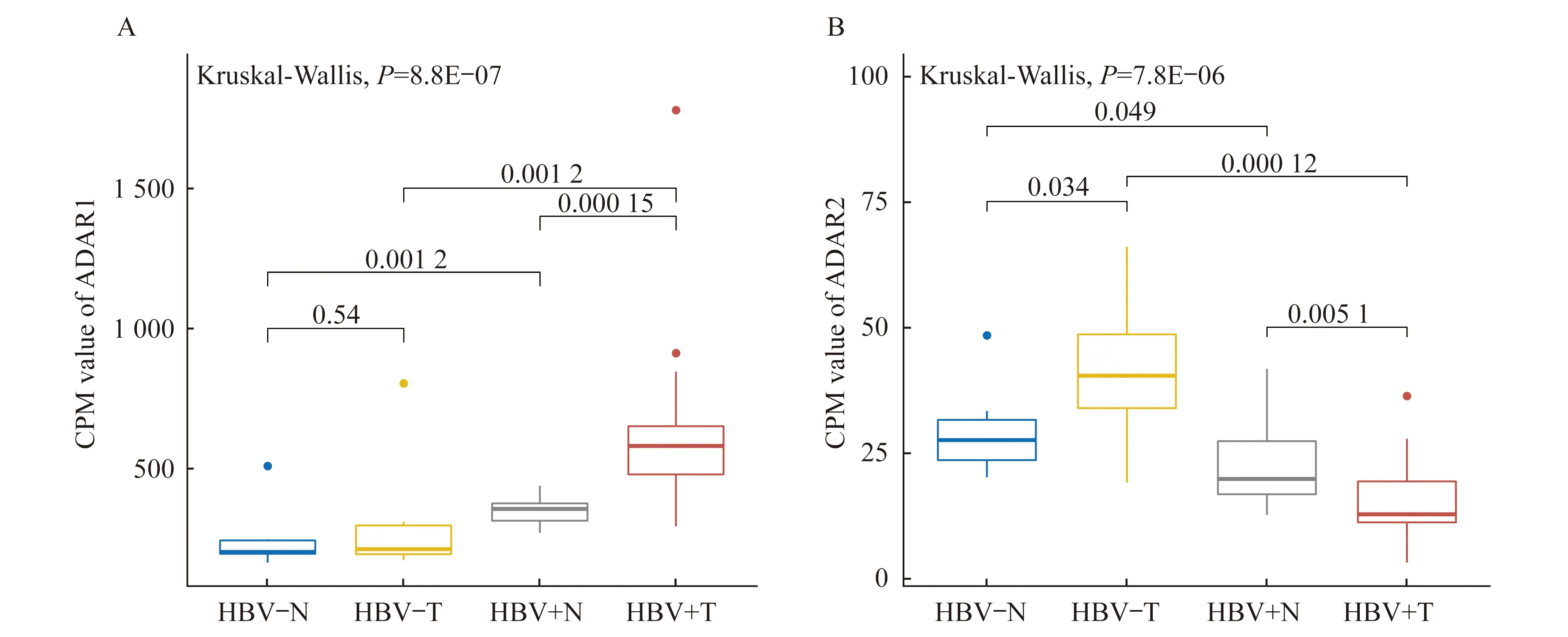
HBV-N:HBV negative and normal tissue;HBV-T:HBV negative and tumor tissue;HBV+N:HBV positive and normal tissue;HBV+T:HBV positive and tumor tissue.CPM:Count-per-million.
图2 不同HBV感染状态下正常组织和癌组织中ADAR1(A)和ADAR2(B)的表达水平
Fig 2 The expression levels of ADAR1 (A) and ADAR2 (B) in normal and tumor tissues in response to HBV infection
A-to-I RESs编辑水平的变化由于ADAR1在催化A-to-I RNA编辑反应中起主要作用,进一步分析ADAR1的上调是否导致编辑水平的显著变化。我们取出每位患者正常组织和癌组织共有的A-to-I RESs后,计算出这些位点的标准化编辑水平。根据共有RESs的编辑水平计算出均值,比较不同感染状况和不同组织来源样本群体的RESs编辑水平变化(图3)。不同感染状态下样本的编辑水平在0.30~0.45,多集中在低于0.5的区域中。虽然HBV感染时ADAR1在两类组织中均显著上升,但仅在癌组织中发现编辑水平上升(P<0.1)。在HBV阳性群体中,癌组织的编辑水平相对于正常组织有所提升(P<0.1),但在HBV阴性群体中则没有类似现象。这说明ADAR1表达变化对癌症样本和HBV感染样本的RESs作用更为明显。
A-to-I RESs所在基因的GO富集情况分析RESs所在基因的GO富集情况,找出在HBV阴性和阳性样本中均出现的GO富集通路(表2):HBV阴性样本的编辑基因显著富集在细胞氨基酸代谢过程通路(P<0.05);HBV阳性样本的编辑基因则富集在蛋白质丝氨酸/苏氨酸激酶活性、转录后调控基因表达及去磷酸化相关信号通路(P<0.05)。这些通路与细胞增殖、基因调控过程相关。这说明HBV感染使细胞增殖并调控基因表达相关的编辑基因活性发生改变,这可能对HCC的发生造成影响。

HBV-N:HBV negative and normal tissue;HBV-T:HBV negative and tumor tissue;HBV+N:HBV positive and normal tissue;HBV+T:HBV positive and tumor tissue.
图3 在不同HBV感染状态下正常和癌症组织中RESs的编辑水平变化
Fig 3 The editing level of RESs in normal and tumor tissuesin response to HBV infection
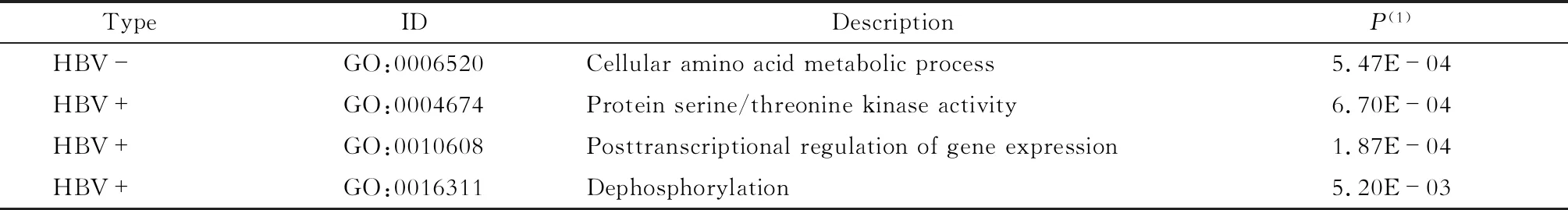
表2 不同HBV感染状态下RESs所在基因的GO富集通路Tab 2 The GO enrichment pathway of genes with RESs in response to HBV infection
HBV-:HBV negative;HBV+:HBV positive.(1)AjustedP.
讨 论
HCC是一种异质性肿瘤,在遗传信息和表观遗传层面表现出复杂多样的变化。在癌组织中,异常的转录后修饰(如RNA编辑),可能导致肿瘤转录组的多样性[23]。RNA编辑在病毒感染过程中同样起着重要作用[7]。目前对HCC患者RNA编辑的研究多为癌组织相对于正常组织的RNA编辑事件变化,对HBV感染后患者RNA编辑的变化研究较少。
我们使用SPRINT软件来检测编辑位点,相对于以往的位点鉴定方法[21],该法能发现更多RES,有利于后续位点的统计分析。通过分析每位患者RES的数目和分布特征,发现两套数据集的RES数目有明显差异,数据测序深度大的样本能检测到更多的RES,这与数据本身测序深度和读数长度有关,因而从位点数量分析HBV感染对患者RNA编辑的影响并不合适。通过分析位点在基因组上的分布,发现不同数据集的样本分布趋势具有较强的一致性,RES大部分落在内含子区域和基因间区域。这与以往研究结果一致[9]。不同HBV感染状态下,正常组织和癌组织中ADAR1表达水平均显著上升。HBV感染后,癌组织中ADAR1表达上调,同时其共同RES的编辑水平也会随之升高,而在未感染HBV群体中则没有类似发现。这说明HBV感染极可能通过上调ADAR1表达,对编辑基因的表达活性产生影响。HBV感染样本的编辑基因显著富集在基因调控和细胞增殖相关通路,说明编辑基因的活性改变可能影响了细胞的正常生长,进而影响HCC的发生。这提示在治疗HBV感染患者的过程中抑制ADAR1的表达对预防HCC的发生可能会起到作用。
本研究收集的数据来自FL-HCC和HCC两套公开数据集。考虑到不同数据集对后续分析的影响,分别对两套数据集在基因组上的分布进行了分析,发现它们在基因功能区的分布是一致的。这在一定程度上表明样本组织来源的不同对RESs分布影响不大。考虑到批次影响,我们在分析基因表达水平时采用标准化基因表达值CPM来衡量ADAR表达水平。在分析RESs的编辑水平时,将正常组织作为背景,选取肿瘤和正常组织共有的RESs来分析,这在一定程度上能够去除由于样本的组织类型差异所造成的编辑水平变化。由于患者年龄、性别和病毒感染等因素也可能对分析造成一定的影响,因此我们在选取数据时尽量选择范围一致的样本。由于公共数据库的数据有限且未感染HBV的临床样本不易获取,本研究收集到的不同HBV感染情况的患者样本数目并不一致。更多未感染HBV的HCC患者样本有助于校正分析过程中的偏差。
总之,本研究利用转录组数据描述HBV阳性和阴性HCC样本A-to-I RNA编辑事件的区别,发现HBV感染上调ADAR1的表达,从而改变宿主编辑事件的活性,这对HCC的发生发展可能有促进作用,也为进一步探索宿主RNA编辑对HBV感染的响应机制提供了线索。
