Recent advances on first-principles modeling for the design of materials in CO2 capture technologies
2019-10-17YueYuanHuabeiYouLuisRicardezSandoval
Yue Yuan ,Huabei You ,Luis Ricardez-Sandoval,*
1 Chemical Engineering Department,Tianjin University,Tianjin 300072,China
2 Chemical Engineering Department,University of Waterloo,200 University Ave W,Waterloo,ON N2L 3G1,Canada
ABSTRACT Novel technologies in consideration of industrial sustainability(IS)are in urgent need to satisfy the increasing demands from the society.IS realizes the production of materials while maintaining environmental and resource sustainability.The chemical materials used in CO2 capture and storage(CCS)technologies play a significant role in the disposal of greenhouse gas emissions coming from large stationary fossil-fired power plants,which breaks the principle of IS and brings severe environmental problems.This study aims at providing a detailed review of first-principles modeling (density functional theory,DFT)of materials in CO2 capture technologies.DFT analysis provides insight into the atomic properties of the studied systems and builds an efficient guidance of the future design of the materials used in CO2 capture technologies.Major materials including oxygen carriers,metal organic frameworks,membranes,zeolites,ionic liquids and some other promising candidates are considered.The computational studies bring the outcomes of the adsorption behaviors,structural characteristics and accurate force fields of the studied materials in short turn-around times at low cost.This review can stimulate the design of novel materials with specific target of CO2 capture and promote the industrial sustainability of fossil fuel combustion technologies.
Keywords:Industrial sustainability CO2 capture Density functional theory Fossil fuels
1.Introduction
With the continuous growth in human population,the demand for food,energy and fundamental living resources such as water has significantly increased.According to Godfray et al.[1],more than 14% of people in the world is still in hunger in this century;also,it is expected that by 2050 a growth in food supply from 70% to 100% will be needed to meet the population’s demands.Industrial activities have also been increased to comply with the global demands.The increase in the industrial production of valuable goods has brought severe environmental problems and the inevitable drop in non-renewable resources such as fossil fuels.Therefore,the sustainability of the environment and resources is becoming a major global concern.In terms of industrial development,the production of valuable materials and products needs to satisfy the increasing population demands;however,they need to do it in a sustainable way such that there is a balance between available resources and manufacture of valuable products.From an engineering perspective,the development of new green technologies can promote engineering sustainability,especially in consideration of industrial sustainability (IS).IS [2]refers to the transformation process including the industry that the overall production scheme contributes to a socially,environmentally and economically sustainable growth.Thus,IS requires industrial and academic practitioners to realize the industrial transformation process by taking into consideration economic,environmental and social aspects that enable the development and deployment of sustainable solutions.New materials and technologies are in urgent need to realize IS.The development and utilization of clean energies have become popular,e.g.wind,nuclear and solar energies[3-5].In addition,the effect and treatment of industrial discharges are major considerations in terms of maintaining IS[6].One aspect critical for the deployment of new (clean) technology in consideration of IS the development,testing and implementation of advanced mathematical frameworks,which play a fundamental role to optimize chemical process sustainability and reduce their footprint on the environment [7,8].Among all the studies in the area of IS,the continuous growth in CO2emissions from fossil fuel combustion has attracted the most attention due to their environmental impact and global sustainability [9,10].
According to the International Energy Agency (IEA) report in 2017[11],fossil fuel accounts for almost 80%of the overall energy supply in 2016,as shown in Fig.1.

Fig.1.Contribution of fossil fuels to the overall energy supply in 2016.
Due to the current energy demands,fossil fuels are still not replaceable by renewables due to their relatively low costs and continuous development of efficient combustion technologies with near-zero emissions [12-14].However,the environmental impact incurred when using fossil fuels during the combustion process has become a global issue that cannot be overlooked.Greenhouse gas effects are one of the most serious problems caused from the combustion of fossil fuels.Accordingly,environmental issues caused by greenhouse emissions do not comply with IS principles in modern manufacturing processes.Among all the greenhouse gas emissions,CO2represents almost 75% of greenhouse gases.One detrimental effect is that this gas has relatively long residence times,i.e.,it remains in the atmosphere for long periods of time[15].Thus,technologies aimed at reducing greenhouse emissions(mainly CO2) are key to curb global warming effects while still using fossil fuels for the large-scale production of power.One approach that can be considered to reduce CO2emissions to the atmosphere is through the deployment of technologies that can capture,store,manage and eventually utilize the CO2produced from large stationary sources such as fossil-fired power plants.The development of efficient CO2capture systems engaged with the continuous production of power from fossil fuels is attractive since it may drive the sustainable development of fossil-fired energy technologies with near-zero emissions.CO2disposal technologies are usually classified as pre-combustion,which reduces carbon capacity of fuels before combustion occurs;postcombustion (flue gases generated after combustion are cleaned);and oxy-fuel combustion,which makes use of pure O2as oxidizer.The process schemes of these CO2capture technologies are described in Fig.2.
Post-combustion is a relatively commercially mature technology which favors low pressure processes.However,the energy penalty of CO2separation while adopting post-combustion technologies (using e.g.amine-based solvents) is from 25% to 40%[16].The post-combustion process normally operates at ambient pressure,low CO2concentrations (usually under 15%) and can often handle large flue gas flow rates.CO2partial pressure is usually less than 0.15 atm [17].Pre-combustion technologies significantly reduce the energy consumptions associated with CO2capture,particularly in systems that operate at high pressures.Low temperatures such as 40°C are preferred to maintain the high efficiency in pre-combustion processes.According to a previous study [18],the energy penalty of the net plant efficiency in IGCC(Integrated Gasification Combined Cycle) plant with precombustion technology is from 7% to 9.5%.Compared to the preand post-combustion technologies,oxy-combustion usually adopts a recirculation process and lowers the energy penalty through the separation of O2from air before combustion.A low mass flux and high CO2concentration of exhaust gas is expected to be separated in oxy-combustion.Despite its benefits,e.g.use of simple separation units to produce a highly pure CO2stream,this technology is still intensive and requires significant energy demands,mostly to separate O2from air.Despite the advances in carbon capture and storage (CCS) technologies,they usually sacrifice part of the overall system energy utilization efficiency to realize the CO2capture [19-21].One key factor that affects the performance of CCS technologies is related to the type materials used to capture the CO2.Therefore,a large and diverse variety of potential materials need to be tested to evaluate its impact on both the efficiency to capture CO2and its corresponding energy costs for recovery.Table 1 summarizes the most frequently used materials in precombustion,post-combustion and oxy-combustion processes.The applications of these materials to different CCS applications will be described in the subsequent sections in this study.

Fig.2.Process scheme of CO2 capture technologies

Table 1 Materials commonly used in CCS technologies
Computer-aided design is key to the development and assessment of promising materials for CO2capture.It has been widely recognized that computer simulations at the atomistic and molecular scales can lead the development of new materials(or the optimal design of existing materials)in short turn-around times and at low cost [43,44]thereby contributing to the sustainability of the environment and industrial processes.In particular,Density Functional Theory (DFT) analysis is a powerful atomistic (firstprinciples) modeling method used to screen out a large number of materials that can be employed for CO2capture.DFT is in the range of the quantum chemical methods and is widely used to predict the properties of chemical structures.To conduct a DFT analysis of different materials such as solvent,adsorption material and oxygen carriers (OCs),different models need to be used.For solid materials,the periodic vacuum slabs of the surface model [23,45]are used to study the surface reactions while the isolated cluster models are usually employed to study particle interactions [46].As for liquid solvents,the continuum solvation model(SMD)developed by Marenich et al.[47]can be used to study the CO2absorption into aqueous solutions [48].DFT calculations are usually conducted at 0 K and under vacuum condition.The DFT energetic results can be adjusted to specific operating conditions using the atomistic thermodynamic models such as the ab initio atomistic thermodynamics [49,50]or through molecular dynamic simulations.DFT analysis can provide insights into elementary reaction kinetics,charge distribution or transfer and microscopic morphological properties of systems under analysis.This modeling method has been proven to be a powerful tool to study the atomic behaviour of promising materials and provide significant guidance for the design of highly efficient (low-cost) materials for CO2capture.
In this work,we aim to review the most recent advances that have employed first-principles modeling (i.e.DFT) to advance our knowledge on the identification and atomistic design of suitable materials for CO2capture.This work focuses on the most common and promising materials used for the different CO2capture technologies such as oxygen carriers(OCs),Metal-Organic Frameworks(MOFs),membranes,zeolites and ionic liquids.Each type of materials will be reviewed in Section 2.Recent advances in other materials such as porous carbons and amine solutions will also be discussed in Section 2.6.A summary of the current state-of-theart in materials for CO2capture will be provided at the end.The existing reports and challenges in DFT analysis of materials used in CO2capture will encourage further computational reports thus promoting IS in our society.
2.Materials in CCS
This section aims to present the recent advances in the commonly used and new chemicals used in the different CO2capture and storage technologies.
2.1.Oxygen carriers (OCs)
Chemical looping combustion (CLC) has attracted attention in the past decades due to its relatively low energy cost compared to other carbon capture and storage technologies[9,51-55].Based on the reaction properties,there are two types of chemical looping schemes that can realize the fuel conversion process.Type I CLC realizes the indirect combustion of fuels by introducing an oxygen carrier (OC),which minimizes the irreversible entropy during the combustion process.As shown in Fig.3(a),in the first reactor(the reducer),metal oxides are reduced to realize the full oxidation of either gas or solid fuels,e.g.syngas or coal.The reduced metal oxides are then transported into the second reactor (the combustor) to be re-oxidized by air thus forming a loop.Through CLC,the combustion process is energy-effective since it simultaneously captures CO2and produces heat thus promoting energy,environmental and ecosystem sustainability.As shown in Fig.3(b),type II CLC also requires two reactors,i.e.a carbonator and a calciner,which includes CO2transport using CO2carriers.In the carbonator,CaO is carbonated by CO2into CaCO3whereas in the calciner the CaCO3undergoes calcination reaction and release CO2at high temperatures which increases the energy regeneration efficiency[56,57].Guo et al.[58]combined experimental and DFT methods to study the metal promoter (Al,Mg,Zr and Na) effect on CaO as OC.That study showed that Zr and Na benefit CO2adsorption while Al and Mg weaken the adsorption.To the authors’knowledge,this is the only study of Ca-based OCs using first-principles (DFT)analysis.
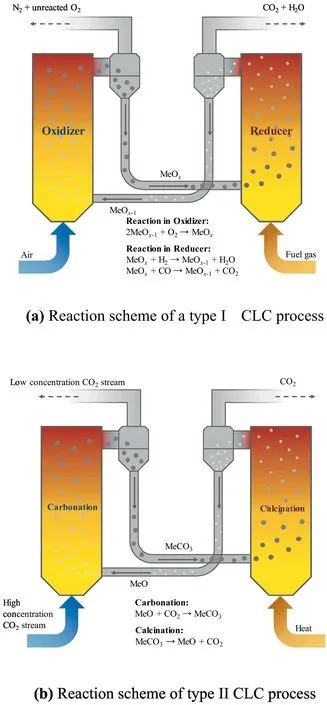
Fig.3.Reaction schemes of CLC processes.(a) Reaction scheme of a type I CLC process.(b) Reaction scheme of type II CLC process.
Oxygen Carrier (OC) development is instrumental for the successful optimization and industrial-scale deployment of CLC technology.This material serves as an oxygen intermediate to combust fuels while energy is supplied by the redox reactions.High reaction reactivity for both the oxidation and the reduction processes is the most desired property for OC materials.In addition,large oxygen storage capacity facilitates fuel conversion.Moreover,to minimize the deactivation of OCs,high mechanical strength and long-term stability under severe operational conditions are desired (2 MPa,around 1000°C [59,60]).Furthermore,OCs should be inexpensive,easy to access and should not lead to serious environmental concerns.Numerous OC materials have been experimentally studied and tested over the past decades[61].Compared to experimental studies,first-principles modeling studies for OCs are relatively limited.DFT,as well as other quantum chemistry methods,have been popular tools used to provide insights into elementary reaction kinetics [62-65].DFT analysis has been mostly employed in this field to study the reactivity of OCs in the redox process.In terms of OC development for CLC applications using DFT analysis,iron-based materials are the most widely studied materials [22,66,67]followed by Cu-based OCs[68].Though NiO-based is one of the most popular OCs in CLC,only a limited number of DFT studies for NiO has been conducted[69,70].Theoretical studies focused on other metals are rather scarce in the literature.
Dong et al.[22]conducted a DFT analysis on Fe2O3as OC using CO as a gas fuel.That study showed that Fe2O3(1102) surface has higher reaction activity with CO than Fe2O3(0001) surface.In another study conducted by the same group,they reported that high index surface Fe2O3(104) performs better than low index(001)surface[71].Carbon deposition[72]and mercury adsorption[73]were also investigated by the same group to assess the impurity effects on the Fe2O3surface.Methane decomposition mechanisms on Fe2O3have been studied using DFT by multiple groups.Different reaction pathways have been established on Fe2O3surfaces [74-76].Due to the complexity of methane decomposition paths,this reaction mechanism is still under debate and will likely be the subject of future research studies in this area.In a different study,Fan et al.[9]considered the oxygen vacancy on α-Fe2O3(001)surface,which has been shown to enhance the partial oxidation of CH4.Other fuel molecules reacting on the Fe2O3surface have been investigated such as carbon atoms or CO [77,78].Those DFT studies reported the reaction kinetics in a solid fuel CLC process.Supports such as Al2O3,ZrO2and MgO have also been studied as the inert support of Fe2O3[66,79,80].ZrO2supported Fe2O3[66]and MgO supported Fe2O3[80]were investigated by Tan et al.Those studies showed that ZrO2could enhance the adsorption of CO while MgO lowered the reaction barriers of CO oxidation compared to the pure Fe2O3OC.
Cu-based OCs are promising materials for the oxygen uncoupling (CLOU) process since O2is released in the reducer.The support effect on the sintering resistance of CuO was studied by Zhao et al.[81];four supports (TiO2,ZrO2,CuAl2O4and MgAl2O4)were studied and all the supports were shown to reduce the energy barriers of the fuel combustion process.Among them,CuAl2O4showed the best performance in the CLOU process.A specially synthesized Cu-based OC with a core(Al2O3)-shell(TiO2)support was studied by Xu and Zhao[82].That study showed that this material could effectively prevent the impurity formation in CLOU process.Studies involving graphene [83]and ZrO2[84]supports have also been reported and have shown that graphene improves the reaction activity of OC with CO whereas ZrO2contributes to the CO oxidation.Liu et al.[85]showed that Zr benefits the oxygen vacancy formation and migration on CuO surface which leads to higher reactivity of CuO as OC.
NiO (001) perfect and defective surfaces have been considered in DFT studies for CLC applications.A report by Cai et al.[23]has shown that the oxygen vacancy on NiO enhanced the O2dissociation in the reduction process.The impurity of CLC (H2S) on NiO(001) perfect and defect surfaces was studied by Guo et al.[86].The results from that DFT study showed that impurity may notably reduce the CO adsorption on the surface.The oxidation process of Ni in CLC was also studied using DFT [87].Other OCs like Mn3O4(001) [88]and CaSO4[89]have also been investigated in CO and CH4conversions,respectively.Those works have provided the elementary reaction kinetics of the studied systems.Theoretical studies on supports of NiO are limited even for the most common Al2O3supported NiO.
Mixed OCs which contain more than one metal elements have attracted much attention as OCs.Studies focusing on perovskites such as BaMnO3and SrMnO3have been reported.Results from those studies have shown that different compositions of those materials have resulted in different reaction activity performances[90,91].Note that Fan et al.[24]reported a DFT analysis combined with experimental data that studied the role of metal oxide support.In that work,the structure of ilmenite(FeTiO3)has been chosen to represent the TiO2-supported iron oxide.That study presents a comprehensive understanding of the TiO2support effect on iron-based OC and provides a representative demonstration of DFT study in this area.Since FeTiO3is the active component of ilmenite while it also contains MgO and Al2O3[92],that study can be used as a basis to develop a new study of trace metal elements in ilmenite as mixed OCs.
In summary,CLC is an energetically favorable process with inherent CO2separation characteristics that can significantly contribute to curb greenhouse gas emissions.To enhance CLC performance,DFT analysis of OCs will provide the fundamental atomistic understanding of the studied systems.However,DFT simulation reports of Ni-based OCs[23]as well as the other materials,natural ores for instance,are limited in the literature.Furthermore,some significant topics such as lattice transfer performance for OCs using DFT analysis have not been reported in the literature.To screen and design promising OCs without too much cost,further DFT-based studies are needed.
2.2.MOFs
Metal-organic frameworks(MOFs)are one of the most promising adsorbents of CO2capture and have become the main focus of research for many experimental and theoretical studies.MOFs are highly ordered crystals containing both metal nodes(metal ions or metal clusters) and organic linkers,which enable MOFs to have a wide variety of properties for multiple applications[93].The diversity of MOFs,different metal sites and different linkers,leads to the need for a large scale screening of these materials using atomistic(DFT)methods.As adsorbents,MOFs are known for their high pore volume,high surface area and internal surface (higher than 7000 m2·g-1) [94].Also,physical and chemical properties of MOFs such as pore size or functional groups can be tailored by choosing different building blocks and synthesis strategies [95].The methods to synthesize MOFs have been the focus of multiple studies in this area since they can potentially tune desirable characteristics for MOFs [93].As for the materials’ strength,structures of MOFs have high robustness,which means that the adsorbed species can be removed to realize the activation of adsorbents while maintaining the crystal structure of MOFs.All the characteristics described above make MOFs promising candidates for CO2capture adsorbents for post-combustion technologies,which have been tested through multiple experimental and simulational studies[96-98].
One area of research in MOF is focused on coordinatively unsaturated sites (CUM),which tend to form reversible bonds with CO2[94].The most popular potentials used to simulate the interactions between CO2and MOFs,i.e.,a combination of Lennard-Jones (LJ)potential and Coulombic potentials[99],cannot accurately predict the adsorption properties since those potentials have been mostly developed for physical adsorption processes.Therefore,firstprinciples-based force field studies are popular to analyze and evaluate the capabilities of MOFs to adsorb CO2[100,101].DFT analysis on MOFs can also provide lattice constants and atomic geometries as well as particle properties during the gas adsorption process,e.g.mechanics of adsorption,particularly for CO2capture[102].Computer-aided design allows efficient and fast screening of MOFs for CO2capture.The aim of this section is to review those recent modeling studies that have considered the adsorption properties of MOFs for CO2such as adsorption,selectivity and impurity effects.Also,a summary of the optimization strategies to improve CO2separation process by MOFs is described.DFT-based force fields,which are constructed to simulate the expected behavior of MOFs in the CO2capture process,will be described at the end of this section.
In the selection of adsorbents to capture CO2,the adsorption properties such as adsorption capacities and CO2selectivity are usually key for the identification of a promising solvent.Ramsahye et al.[103]calculated the charge distribution of the MIL-53 by DFT analysis.Periodic models as well as cluster models of MIL-53 were employed and have shown to achieve good agreement with structures observed from experiments.The charge distribution predicted from DFT simulations was used to simulate a grand canonical Monte Carlo (GCMC) model,which is the most commonly used simulation method in this area.Then the adsorption capacity,as well as selectivity of CO2,were estimated.That work showed that the Mulliken charges from periodic models are more transferable between the two changing structures of MIL-53 due to its flexibility,which would provide a better prediction for the entire adsorption process.Hu et al.[104]showed that Zr-BFDC(O-heterocyclic ligand contained)performed the best among three studied UiO-type of MOFs.MOFs have been simulated in numerous studies and shown to be promising CO2adsorbents with high adsorption capacity and easy to regenerate.Borges et al.[105]reported a study focused on selectivity of CO2capture among H2,N2,CH4and CO.That study indicated that MIL-160 was a promising adsorbent of selective CO2adsorption.The adsorption mechanism(i.e.the binding energies of CO2) was obtained from DFT analysis.The results from that work showed that CO2tends to be adsorbed on the Zr-metal cluster.Liu et al.reported[106]that the side position where the CO2is closer to the hydrogen side of the organic linker is the most favored site among multiple configurations.DFT has been shown to be an effective tool for the prediction of adsorption mechanism in MOF-contained systems.Impurity effects on the CO2adsorption process can also be obtained from DFT simulations.Yu et al.[107]simulated the CO2adsorption using Mg-MOF-74 in a system that contained water.The water-coordinated MOF has lower binding energy with CO2,which leads to a decrease in the adsorption capacity of CO2.Regarding the functional group effects,Torrisi et al.[97]reported four functionalized MIL-53 containing different groups (OH-,COOH-,NH2-and CH3-).The results reported that each functional group has different favored adsorption sites and bonding energies.Functional groups (especially OH-and COOH-) have enhanced the adsorption of CO2by MIL-53 in consideration of capacity of CO2and the selectivity of CO2over CH4.
The computational studies mentioned above provide guidelines for the optimization of MOFs or even the design of new MOFs for CO2capture.Unsaturated sites,polar functional groups,alkylamine incorporation and pore size can contribute to CO2capture with MOFs [94].Nazarian et al.[108]analyzed 879 MOFs whose structures were optimized using DFT analysis.In some cases,the computationally optimized structures make the CO2adsorption significantly different from the experimental results;the major source of discrepancy was due to the use of force fields that were not able capture the key characteristics of the system;therefore,the development of accurate force fields for those systems is indeed needed to further advance the design of new MOFs materials.In terms of force field development for MOFs,DFT calculations can optimize the structures of the studied materials such as the surface model or the isolated cluster configurations.Also,the interactions and distances between the particles can be obtained by DFT analysis as well as the energetic and electronic properties of the configurations.These results are then used in the specific force field models such the 12-6 Lennard-Jones potential.Grand Canonical Monte Carlo (GCMC) simulations can be used to validate the force field models by comparing the GCMC predictions with experimental observations [109].Especially for the MOFs with open metal sites,general force fields such as UFF (Universal force field)[110]cannot accurately simulate the adsorption behavior since they cannot predict the strong interactions present between CO2and the adsorbent containing open sites.As mentioned above,CUM contained MOFs has been widely studied using DFT analysis given that the open metal site has the potential to form a chemical bond with CO2.Haldoupis et al.[111]reported that the favorable adsorption sites of CO2are the existing open metal sites of the studied MMOF-74(M stands for Mn,Co,Ni,Cu).Also,they parametrized the interactions between CO2and the unsaturated sites using the DFTderived interaction energy.The developed force fields were able to predict consistent adsorption isotherms that agree well with previous experimental results.Poloni et al.[112]studied MOF-74 (Ca and Mg) as well as BTT-type MOFs containing unsaturated sites(BTT stands for 1,3,5-benzenetristetrazolate).That study showed that binding energies are highly sensitive to the existing divalent cations in BTT;similarly,the open V and Ti sites favored the CO2adsorption in M-MOF-74.
Another research avenue in MOFs is the simulation of breathing MOFs,i.e.MOFs with flexible structures like MOF-5.Adequate force fields that can predict flexible MOFs are currently under investigation [113].Zhao et al.[25]performed a DFT analysis to obtain the angle bending parameters around Cu of Cu-BTC (Cu3(BTC)2with BTC (benzene-1,3,5-tricarboxylate),which is a breathing MOF.The new force field,which was developed to describe the breathing MOF,Cu-BTC,combined parameters estimated from DFT and slightly modified existing parameters,i.e.the intramolecular force constants from a widely used CVFF (Consistent Valence Force-Field) [114,115].The new force field is evaluated by its prediction of Cu-BTC crystal structures,adsorption behavior,etc.According to the comparison of the experimental outcomes and simulated results,the newly developed force field can give an accurate prediction of the properties of Cu-BTC such as the crystal structure,vibrational properties and the adsorption behavior.
DFT analysis can provide the characteristics of MOFs such as lattice parameters of MOFs,which can lay a solid foundation for future structure-relation studies of CO2capture using MOFs.Also,the development of new force fields improve the accuracy of the simulational outcomes and extend the systems that can be explored using DFT analysis.
2.3.Membranes
The application of membranes in CO2capture mainly includes H2/CO2separation for pre-combustion,CO2/N2separation for post-combustion and O2/N2separation for oxy-fuel combustion.Membranes act as filters in the separation process since they only allow targeted components in the gas mixtures to permeate through,while leaving the rest of the species behind,which produces a pure stream of the desired gas component.Membranebased technology has shown great potential in CCS since it does not require significant investment in large-scale power plant facilities.Also,this technology is not energy intensive like the traditional solvent-based technologies,which require large amounts of heat to regenerate the solvent.Moreover,membranes only need to be replaced when they reach their lifespan.Depending on their specific configuration,membranes can also be designed to suit the different needs for the separation processes.
The design of a membrane for CO2capture is mostly driven by the permeability and selectivity of the membrane.Permeability represents the gas permeation flux per unit pressure,whereas selectivity denotes the ratio of the permeabilities among different gas species.Membranes used for CO2capture are typically classified as:inorganic frameworks (e.g.metallic,carbon,silica,zeolite and metal-organic frameworks [116];and organic frameworks,which mainly includes cellulose acetate and polymers [117].
Considering the number of different frameworks and possible configurations that can be employed to build a membrane,measuring the performance of the different membrane candidates under a wide range of operating conditions in a laboratory is not only time-consuming,but also expensive.On the other hand,computational simulations can be used for screening and identification of the most promising structures for different applications.In what follows,we will discuss the first-principles calculation studies on membranes that are H2-selective,CO2-selective,and N2-selective,respectively.
H2-selective membranes are mainly used in pre-combustion CO2capture process to separate a mixture of H2and CO2at high pressures and temperatures.Metal membranes are to a certain extent ideal for this type of processes because of their potential infinite H2selectivity [118].Hydrogen purification membranes made of Palladium (Pd) and its alloys have been heavily studied over the past 50 years [28].The main drawback of pure Pd-based membranes is that they are prone to surface contamination and H2-induced embrittlement at temperatures below 300°C.On the other hand,Pd-alloy membranes have the potential to resist surface poisoning while maintaining desirable properties of pure Pd membrane.Kamakoti et al.[119]used plane wave DFT calculations to examine the binding energy and diffusion activation energies for dilute interstitial H in pure Pd,bcc and fcc CuPd alloys with~50 at.%Pd.The dilute interstitial hydrogen loading was approximated by placing a single H atom on each of the compound’s computational supercells.The diffusion activation energy was calculated using harmonic transition state theory(TST)with zero-point corrections.Results from this study have shown that H atom has the strongest binding energy with pure Pd,which also has the smallest net activation energy for H diffusion.As an indicator of the calculation accuracy,the predicted H diffusion activation energy in pure Pd(0.24 eV)was found to be in excellent agreement with experimental results(0.23 eV).Ling et al.[120]applied DFT-based calculation to evaluate the interactions between H2and a wide range of compositions of PdCuAg alloy membranes.DFT calculation was employed to obtain the hydrogen solubility,diffusivity,and permeability of each composition examined.A contour map was generated as a function of the alloy composition based on a large collection of DFT results.That study concluded that the solubility of hydrogen increases with Ag concentration while this variation can also cause a decrease in the diffusivity.Specific compositions that have a higher permeability than pure Pd have also been identified.Chandrasekhar et al.[121]calculated hydrogen solubility for 78 Pd-based binary intermetallics and used this information as a screening parameter to generate a shorter list of candidates to study hydrogen permeability.The study revealed that no material has higher hydrogen permeability than pure Pd.However,their work significantly increases the set of materials for which hydrogen permeability is known.
Carbon-based membranes separate H2from gas mixtures by molecular sieving.Zhang et al.[122]calculated the H2permeability and selectivity of sp-sp2hybridized carbon allotrope membranes with different natural pores,i.e.,graphyne,graphdiyne,and rhombic-graphyne.Their calculations showed that these properties are highly dependent on the pore sizes and membrane shapes.Among all the structures,rhombic-graphyne showed the largest discrepancy in penetration energy barrier for different gas molecules (0.54,1.55,1.73,and 3.00 eV for H2,CO,N2,and CH4respectively);this promotes high H2selectivity (>1016) while maintaining a reasonable H2permeability.Wang et al.proposed four differently modified graphene membranes with Hdeactivated pores (HP6,HP10,HP13,and HP16) and investigated their abilities to separate H2from gas mixtures (see Fig.4) [29].In that study,DFT-based calculations were performed to obtain the permeation energy barriers of each gas species (H2,CO,N2,and CH4).It was found that H2has the lowest permeation energy barriers on all four pore structures studied.Among all membranes examined,HP6 membrane exhibited the highest H2selectivity followed by HP10.However,large permeation energy barriers of H2have also been observed on HP6 and HP10 membranes(13.64 kcal·mol-1for HP6 and 7.05 kcal·mol-1for HP10,1 cal=4.1868 J);these results suggest low gas permeabilities.As for HP13 and HP16,the H2permeation energy barriers are very small and can result in high permeabilities (0.52 kcal·mol-1on HP13 and 0.03 kcal·mol-1on HP16).However,the calculated energy barriers of CO on HP13 (1.06 kcal·mol-1) and HP16(0.94 kcal·mol-1) are not significantly higher than those obtained for H2,which can lead to poor selectivity of H2over CO.On the other hand,HP13 and HP16 can be used to separate H2from CO2and CH4with the selectivity of 4.6×103(HP13) and 4.3×102(HP16) for H2/CO2mixture;and the selectivity of 5.4×108(HP13) and 1.1×104(HP16) for H2/CH4mixture.These results indicate that H-deactivated graphene could be a promising material for H2-selective membranes.

Fig.4.CO2 and H2 penetrate through HP13 membrane.
CO2-selective membranes are mainly used in post-combustion CO2capture process to separate a mixture of N2and CO2.Cazorla et al.[123]calculated the CO2selectivity and capacity of the Calcium-decorated carbon nanostructures using DFT analysis.In their work,the Perdew-Burke-Ernzerhof generalized gradient approximation (PBE-GGA) for the exchange-correlation energy was employed.The calculated binding energy of N2is much smaller than CO2on the Ca-doped surface (Ebind,N2=-0.645 eV and Ebind,CO2=-2.731 eV on XCa=12.4% graphene surface).Also,the formation of calcium carbonate (CaCO3) promoted CO2selectivity.These results suggest that Ca-decorated carbon nanostructure membranes are suitable for their applications in post-combustion technologies.Ostwal et al.[124]used DFT calculations to investigate the means of CO2hopping on the 3-aminopropyl-triethoxy silane (APTS) functionalized silica membrane.The binding energy of CO2adsorption (15.5 kcal·mol-1) and the activation energy of CO2diffusion/hopping from one amine group to another of APTS(7.2 kcal·mol-1)were in good agreement with experimental results(14.33-21.5 kcal·mol-1and 8.0 kcal·mol-1,respectively).Therefore,CO2molecules are likely to follow the proposed mechanism when they interact with the APTS membrane.Wu et al.[125]also performed DFT-based simulations to investigate the mechanism of the fluorine-modified porous graphene membrane for CO2/N2separation.Their result showed that the pore-22 graphene (with 22 carbon atoms drilled out) has small diffusion energy barriers for both CO2and N2,which leads to a low selectivity.However,once the fluorine has been modified,the diffusion barrier for CO2decreased to 0.029 eV,while the diffusion barrier for N2increased to 0.116 eV.This difference suggests that CO2will pass through the membrane much easier than N2,thus enhancing the selectivity of CO2/N2mixture.Watanaba et al.[30]predicted the capability of a MOF material Cu(hfipbb)(H2hfibpp)0.5(H2hfipbb=4,4′-(hexafluor oisopropylidene)-bis-(benzoic acid) for membrane-based CO2/CH4separation.In their work,DFT calculations were used to confirm the significant difference between the diffusion energy barriers of CO2and CH4.The diffusion activation energies of CO2and CH4in the pore of Cu(hfipbb)(H2hfibpp)0.5were calculated using TST.According to their calculations,the activation energies for CH4and CO2are 45 kJ·mol-1and 16 kJ·mol-1respectively,which justifies the high CO2selectivity exhibited by this material.DFT calculations has also been used to provide geometry optimized structures [126]or force constants and atomic charges that can be used to develop force fields (FFs) for molecular dynamic simulations [127](see Section 2.2).
The concept of a N2-selective membrane stems from the development of H2-selective metallic membranes.Rochana et al.[128]studied the molecular dissociative adsorption of nitrogen and potential subsequent atomic diffusion within the Vanadium (110)surface and alloys with Ruthenium (Ru).That report showed that the binding energy of N2and atomic nitrogen bond on the V(110)surface is stronger than that of the Fe surface,which is traditionally used as the catalyst for ammonia synthesis.However,compared to Fe,N2has higher activation energy barrier for the dissociation process(0.4 eV)and the subsequent subsurface diffusion process (1.4 eV).As a result,the authors of that study concluded that nitrogen transport through a pure V membrane may be difficult to achieve.V-Ru alloys,on the other hand,may have the potential to be used as membrane materials,i.e.,DFT simulations performed in that study showed that the presence of Ru weakens the adsorption of nitrogen in the first subsurface layer,which enhances the diffusion of atomic nitrogen.Wang et al.[129]revealed that the graphene membrane with nanopores formed by replacing 13 C atoms with H atoms(H-pore-13)can efficiently separate N2from CO2despite the fact that the size of the pore(0.406 nm) is larger than the size of both N2and CO2molecules.The authors further studied this phenomenon using dispersioncorrected DFT by portraying the potential energy profiles of N2and CO2molecules penetrating through the pore.The profile showed that CO2and N2molecules interact with the pore most strongly at 0.2 nm and 0.16 nm above the surface with interaction energies of 0.24 eV and 0.14 eV,respectively.Moreover,the energy barriers to pass through the pore are 0.19 eV for CO2molecule and 0.05 eV for N2molecule.Hence,it is more difficult for CO2to permeate the membrane compared with N2.Similar methods were also used to examine the N2selectivity of the poly-(triazine imide)(PTI) membrane [130].The computed energy barriers for CO2,N2and H2O passing through the PTI membrane are 0.262,0.118,and 0.164 eV respectively,which suggests that PTI is also a promising material for N2/CO2separating membranes.
DFT analysis has been successfully employed to theoretically investigate and predict the applicability of a wide range of materials in membrane technology.More studies are expected in this field in order to provide guidance to the experimental work and allow experimental scientists to make decisions faster thus enabling the rapid development of new and optimized membrane materials for CO2capture technologies.
2.4.Zeolites
Zeolites are crystallized porous aluminosilicates that contain exchangeable cations (Li+,Na+,Ca2+,etc.).Due to the cages and channels as well as the changeable cations,zeolites have been widely used in pre-combustion and post-combustion CO2capture applications.The diversity of zeolites makes these materials attractive for the CO2adsorption process.For example,the topologies,the number of exchangeable cations in the cavities,pore sizes and Si/Al ratio are important criteria that need to be considered in the selection of zeolites for CO2adsorption [131,132].
DFT analysis of zeolites as CO2adsorbents can predict the adsorption behavior such as adsorption capacity,favored sites and selective adsorption among gaseous mixtures.Zukal et al.[31]reported the CO2adsorption in Na-A zeolites using a periodic slab model to provide accurate predictions of the dispersion interactions.That study showed that simulation results agree well with the experimental outcomes and CO2tends to be stable when it interacts with 3 Na+cations simultaneously.Thang et al.[133]studied the faujasite-type zeolites using the DFT/CC(coupled cluster)method[134].Compared to results reported from other simulation methods,a better prediction of the interaction energy through DFT/CC calculations was obtained in that study,which has also been shown to agree well with experimental results.Cation site effects have been studied by Nachtigall et al.[32].In that study,periodic DFT calculations indicate that dual or multiple cation sites tend to adsorb gas molecules rather than single cation sites.As for CO2in mixtures,the selective adsorption of CO2over N2(exhaust gas composition) is studied with a NaKA zeolite (Na-A type zeolite with partial substitution of Na+by K+) [46].Both the isolated cluster and the periodic model were adopted in that study and have shown that it is the high carbonate forming at the K+ion positions in the 8R windows,which enhances the selectivity towards CO2adsorption.Another focus of selective adsorption on CO2is with natural gas (i.e.a mixture of CO2and CH4).Fischer et al.[135]conducted the dispersion-corrected DFT calculations to estimate the interactions of the adsorbed molecules (CO2,N2and CH4)with different synthetic zeolites.The results from that work showed that the difference in the binding energy between zeolite-CO2and zeolite-CH4 in K+or Rb+containing adsorbents is significant for the selective adsorption of CO2.Compared to other CO2capture materials such as MOFs,DFT studies using zeolites are limited despite the fact that atomistic computational methods have shown to be efficient to predict CO2adsorption phenomena.
2.5.Ionic liquids
Ionic liquids(ILs)are considered promising candidates in industrial processes for conventional volatile compounds separation and storage [136].ILs are salts with melting points lower than 100°C[137].This type of materials often present excellent thermal stability,good solubility as well as the possibility of ionic and composition substitutions,which make them attractive for different industrial applications including CO2capture.ILs also allow the combination of different ions,which offers the flexibility to tune materials for further improvement.This makes ILs particularly attractive since they offer the potential to be designed for specific purposes.Compared with experimental reports,DFT analysis is an efficient and inexpensive approach that can be employed to screen a large number of potential ILs to build up specific structure-property relationships.ILs are popular in the area of CO2capture due to its experimentally reported high CO2capture capacities and its desirable chemical or physical properties [34,138,139].
DFT analysis on ILs applied for CO2capture is usually combined with molecular dynamic simulations (MD).DFT simulations predict the charge distribution and charge transfer,structural and energetic properties of short-range intermolecular interactions within isolated ions,ion-ion and ion-CO2mixtures [140].On the other hand,MD predicts the dynamic properties of the systems such as the diffusivity of CO2confined at specific pressures and temperatures.Aparicio et al.[141]reported the microscopic structures and intermolecular forces of choline benzoate and choline salicylate ILs in the pure state and also in the CO2absorption process.The studied CO2-ion pairs show strong interactions with benzoate anion.The volume increase of the studied ions,cholinium benzoate ([BE][CH]) and cholinium salicylate ([SA][CH]),is apparently larger than other ions such as lactate [142].The mechanism of CO2absorption on the studied ILs as well as the CO2effects are provided.That study reported that there is no overlap in the system containing the studied ions and CO2.Also,the CO2absorption increases the molecular mobility and viscosity of the heterogeneous systems.Shaikh et al.[33]used DFT analysis and MD simulations to predict CO2absorption on amino acid ionic liquids(AAILs).That study showed that glycinate([GLY])adsorbs CO2better than 1,1,1-trimethylhydrazinium ([aN111]) though [GLY]increased the liquid viscosity.The balance of high CO2capture capacity and low viscosity should therefore be taken into account for AAILs.Steckel[137]studied the interaction of CO2with acetate ion.Multiple DFT functionals were evaluated and have shown that they were not able to accurately reproduce all the geometries and energetic calculations obtained from coupled-cluster singles and doubles (CCSD) optimizations;on the other hand,the rangeseparated hybrid meta-GGA [143](M11) and nonlocal (VV10[144]and vdwDF10[145])functionals lead to better results among all the studied DFT methods.That study gives guidance of DFTbased MD calculations and the parametrization of the force fields based on DFT analysis.Damas et al.[146]used DFT to evaluate the interactions of cations and anions derived from ionic liquids with CO2,SO2and H2S.The results suggest that IL-based anions are more likely to interact with CO2when the cation-anion interactions are weaker.That study provides a comprehensive mechanism of the studied system and more absorption species since it extends on a previous report[147]that only considered CO2capture adopting the 1-ethyl-3methylimidazolium (C2mim+) and bis(trifluorosulfonyl)imide (Tf2N-).Garcia et al.[148]screened different density functional methods to study CO2capture by ILs.Binding energies of gas-ILs of a set of 54 ILs were estimated.The results show that ωB97XD [149]can simulate the long-range corrections to provide suitable charge transfer considering the dispersion corrections through DFT-D2 approach [150].The resulting correlations between CO2and ILs are key to optimize the design of ILs for CO2capture.To mimic the interactions in consideration of both physical and chemical absorption,a new force field was developed in CO2absorption on ILs.Zhang et al.[35]developed a ReaxFF force field based on CO2capture by tetrabutylphosphonium glycinate,[P(C4)4][Gly].The resulting force field is used in NPT ensemble MD simulations to predict the equilibrium density at 300 K and 101.325 kPa.Multiple sets of interactions are considered such as periodic DFT simulations of the reaction pathways between CO2and anion in the condensed phase,gas-phase CO2-anion interactions.The ReaxFF-based MD simulation predicts the equilibrium density at 300 K,which is within about 1% of the experimental value.Mondal et al.[151]provided force field parameters for imidazolium cation based ILs combining anions of acetate,thiocyanate and dicyanamide.The DFT-derived fractional charges were used to modify the CLaP force field [152-154]and build a transferable force field.The improved force field was used in MD simulations and showed that the intermolecular structure and dynamics agree well with experimental results.That work showed that the scheme used to define the non-bonded and torsional interaction parameters by the partial charges can be used to establish transferable force fields in this area.
The DFT studies of ILs discussed above show that simulations are powerful tools to screen this highly diverse and tunable material based on the specific targets pursued for CO2absorption.However,some aspects of greenhouse emission disposal are still under debate using ILs.Further research on absorption selectivity of CO2over impurities is therefore needed to further support the development of this material for CO2capture applications.
2.6.Additional materials
The materials discussed in the previous sections represent the most commonly used materials considered for CO2capture.First principles(DFT)studies on other materials that have shown potential as attractive CO2adsorbents.These materials are discussed next.
Porous carbons stand out in the area of CO2adsorption due to their relatively low prices compared to the other materials such as MOFs [16].In addition,a variety of allotropes of carbon such as nanotubes or graphenes allow the specific applications of porous carbons.Mo et al.[37]conducted a functionalized carbon study to analyze CO2adsorption.The N-containing molecular segment models of coal (2-methylpyridine,C13H9N,C23H12N,and N-doped graphenes) was evaluated by CO2uptakes.That study found that the DFT method of BLYP-D3 combined with a 6-311++G(d,p)basis set can accurately simulate the interactions in the studied particles such as 2-methylpyridine,C13H9N and C23H12N,which improves the accuracy of the calculations of CO2adsorption in the system containing similar compositions.Jiao et al.[36]showed that the addition of pyridinic-nitrogen in nanotubes enhances CO2adsorption strength with varying charge states.That study showed that reversible CO2adsorption (i.e.desorption) can be controlled through the injection of electrons into the nanotubes.Then,the idea of switching the charge-carrying states of the nanotubes becomes a new scheme for CO2uptakes.As for fullerene (C60),Gao et al.[155]used DFT analysis to evaluate the potential application of fullerene of CO2adsorption.Their results showed that calcium decorated fullerene have the potential to adsorb 16 CO2molecules in a unit cluster with 4 decorated Ca atoms.Graphene has also been studied for CO2adsorption using DFT calculations.Rad et al.[38]provided microscopic information of the CO2uptakes by Al-doped graphene such as the adsorption energy and electronic states.The outcomes of that study showed the potential of functionalized graphene materials as CO2adsorbents.Another porous carbon materials that have been studied are mesoporous silica nanoparticles such as SBA-15 [39]or MCM-41 [40].Zhuo et al.[156]simulated the CO2and N2adsorption on mimetic MCM-41.DFT analysis of this system showed that CO2has preferable adsorption sites over N2on the materials’ surface.MCM-41 also has higher adsorption selectivity of CO2over N2.As for SBA-15,Zukal et al.[157]evaluated the functionalized SBA-15 with 3-aminopropyl-trimethoxysilane using the CO2adsorption energy distribution obtained from non-local DFT calculations.That study revealed that amine ligands on the surface seem to enhance CO2capture on the studied material.The CO2adsorption capacity of the studied material is highly related to the amount of amine groups.Especially,the tethered amine ligands react strongly with CO2to forma carbamate.The high isosteric heats of adsorption indicate that strong interactions between CO2and amine groups exist.Amine-based solvents,e.g.monoethanolamine (MEA),appears to be the most promising and widely used materials in amine scrubbing post-combustion technology to capture CO2[42,43,158-162].Yamada et al.[48]developed a continuum solvation model(SMD)[47]in DFT analysis to study the CO2absorption in an aqueous solution of 2-amino-2methyl-1-propanol (AMP).SMD is popular to predict solvation free energies of neutral and ionic in the solutions.The established pathway of CO2absorption is a two-step mechanism via a zwitterion intermediate.The carbamate forms fast and decomposes reversibly.Except for the materials mentioned in this section,an increasing number of innovative materials are considered in the area of CO2capture,such as new polymers [163],zinc orthogermanate [164],perovskites [165],metal oxides [166,167]and hydrotalcite [168].Polymers have attracted much attention recently due to their structural tunability[163].In particular,Zinc orthogermanate (Zn2GeO4) is a polymer ternary oxide material that has shown to be promising to capture CO2in photoreduction.This material plays a vital role to improve the reduction of CO2into renewable hydrocarbon fuel as a photo catalyst[164].In addition,perovskites can be regarded as composite OCs that have high activities and also favor ionic and electronic migration during the redox reactions[91].Unlike other OC materials,perovskites allow relatively low energy for oxygen anions and electron transport from the bulk to the active surface.The urgent need for high efficient CO2disposal technologies encourages practitioners to explore new and novel materials with suitable properties for CO2capture.
3.Summary
This review has presented the recent contributions and advances on first-principles (DFT) modeling applied to the design of materials suitable for CO2capture technologies.Testing and validation of computer-aided design materials using experiments will shed light into the development of new materials that can eventually promote the sustainable production of power using fossil-fired systems.The oxygen carrier (OC) used in CLC has attracted much attention due to its energy efficiency since it makes use of oxycombustion process through a 2-reactor scheme.The application of OCs in different systems still requires more research in DFT analysis to provide microscopic structure-property relations,which will guide the design of materials depending on the specific CO2capture targets.MOFs and ILs are both favored by their tunable properties since in principle they can be designed to satisfy a specific separation target requirement.The focus of MOFs in the application of CO2capture is on its unsaturated sites as well as the breathing property of some of the specific MOFs.On the other hand,zeolites,porous carbons,amine-based solutions are relatively mature candidates in the area of CO2disposal,which are currently applied in industry.However,insights on the mechanisms and microscopic structures of the systems using these relatively traditional materials are still lacking in the literature.DFT methods can significantly enhance the application of these materials since they can reveal properties that can be exploited for CO2capture.Computational modeling allows efficient screening of a large set of materials and can accelerate the development of novel materials that can support fossil-fired power production under near zero emissions.Therefore,modeling studies in this area can drive the design and optimization of new materials with properties ideally suited for CO2capture thus promoting the sustainability of new and efficient CO2capture technologies.
Acknowledgements
The authors gratefully acknowledge the support provided by the Chinese Scholarship Council to develop this research.
杂志排行

Chinese Journal of Chemical Engineering的其它文章
- Microencapsulated ammonium polyphosphate by polyurethane with segment of dipentaerythritol and its application in flame retardant polypropylene☆
- Distributed control and optimization of process system networks:A review and perspective☆
- Heat exchanger network synthesis integrated with flexibility and controllability☆
- Synthesis of flexible heat exchanger networks:A review☆
- Simulation and heat exchanger network designs for a novel single-column cryogenic air separation process☆
- A review of extractive distillation from an azeotropic phenomenon for dynamic control☆