氮取代对环状烃在H-FAU分子筛中吸附性能的影响
2019-09-18于双林周立元刘熠斌
姚 远,于双林,党 宇,周立元,刘熠斌
(1.中国石油石油化工研究院,北京102206;2.中国石油大学(华东)化学工程学院)
催化裂化是重油轻质化的重要手段。石油资源的不断重质化、劣质化使得催化裂化过程被迫掺炼低品质原料,如常压渣油、焦化蜡油(CGO)等[1-2]。这类原料中的氮化物尤其是碱性氮化物含量较高,容易导致固体酸催化剂(分子筛)发生中毒失活,给催化裂化过程带来不利影响[3-4]。
目前对催化裂化催化剂氮中毒机理仍无统一认识,一般认为主要有3种作用机制,即酸碱中和理论[5-6]、竞争吸附理论[7-9]和诱导效应[10]。3种中毒理论均认为催化剂中毒深度与氮化物碱性密切相关,这是由于强碱性氮化物通常具有更强的吸附能力和诱导效应,更容易从催化剂表面B酸中心获得H+或者向L酸中心提供孤对电子。Corma等[10]在喹啉、吡啶和2,6-二甲基吡啶对正庚烷在FAU分子筛上裂化性能的研究中发现,碱性氮化物的中毒是由诱导效应导致,并且诱导效应随氮化物碱性的增强而增大。熊秀章等[11]采用蒙特卡罗(GCMC)方法探究了几种碱性不同的吡啶衍生物在FAU分子筛上的吸附行为,结果表明吸附能大小随氮化物碱性的增加而增大。刘银东等[12]以焦化蜡油为原料考察了竞争吸附对催化裂化反应的影响,发现原料中的氮化物具有强吸附能力,并且碱性较强的氮化物在反应过程中的快速吸附是导致催化剂生焦失活的重要原因。但是也有研究表明氮化物的毒性与其碱性强弱并没有一致性[13]。这是由于氮化物的碱性通常在常温下测量,而在催化裂化的高温条件下碱性大小与常温条件下结果相去甚远。
截至目前,不同碱性氮化物对催化剂影响程度的定量研究仍未见报道。本研究选取碱性不同的吡啶、哌啶,以及结构与其相近的苯、环己烷为模型化合物,采用GCMC和密度泛函(DFT)方法于温度298 K下考察上述4种模型化合物在H-FAU分子筛上的吸附行为,计算得到吸附等温线、吸附热曲线、吸附质分布和电子密度变化等结果,对比分析氮取代对环状烃吸附性能的影响,为氮化物对反应过程影响机制的研究提供理论指导。
1 计算模型与方法
1.1 分子筛模型
GCMC计算过程中使用的H-FAU分子筛晶体化学数据为:H28O384Al28Si164,立方晶系,空间群为Fd-3m;晶胞参数为a=b=c=2.503 nm,α=β=γ=90°;主要结构是八面沸石笼,笼口孔径为0.74 nm×0.74 nm;分子筛硅铝比为5.86,与实际使用硅铝比一致。根据文献[14-15]对分子筛中各原子电荷指定如下:Si(+0.89e),Al(+0.73e),O(-0.33e),H(+0.083e)。计算中使用的晶胞组成为1×1×1,并在X,Y,Z3个方向添加周期性条件。对扩大后的超胞进行模拟,未发现明显的尺寸效应,表明上述晶胞大小合适。最终构建的H-FAU分子筛模型及其孔道结构特征如图1所示。
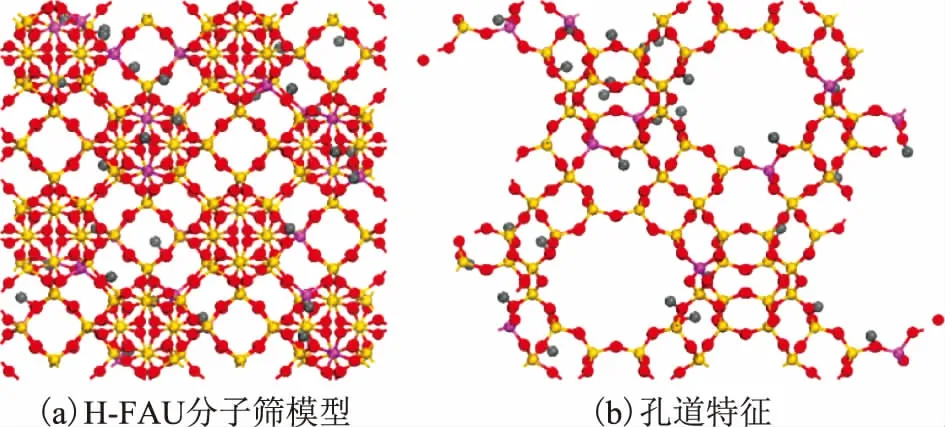
图1 H-FAU分子筛模型以及孔道特征●—Al; ●—Si; ●—O; ●—H
DFT计算主要是为了考察4种模型化合物在H-FAU分子筛酸性位上的化学吸附。受计算条件限制,模拟中采用40T的团簇模型(见图2),包括超笼的十二元环和SOD笼的六元环,活性位点H原子落在两个超笼之间的十二元环上[16-17]。Wang Yajun等[18]采用该模型计算了四氢萘在酸性位上的吸附能,计算结果与实验值一致,说明该团簇模型能够准确地描述FAU分子筛孔道和酸性位的特征。
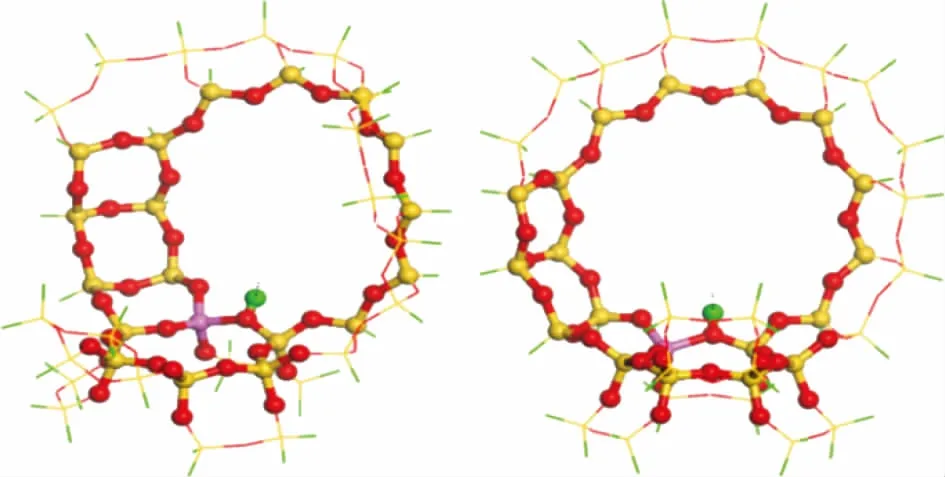
图2 H-FAU分子筛的40T团簇模型●—Al; ●—Si; ●—O; ●—H
1.2 客体分子模型
吡啶、哌啶、苯、环己烷4种分子的结构示意如图3所示,三维尺寸见表1。从表1可以看出,4种分子的最小截面尺寸分别为:0.72 nm×0.36 nm(吡啶)、0.73 nm×0.54 nm(哌啶)、0.72 nm×0.32 nm(苯)、0.69 nm×0.52 nm(环己烷),均小于FAU分子筛超笼的笼口直径(0.74 nm×0.74 nm),因此理论上这4种分子可以扩散至FAU分子筛孔道中。
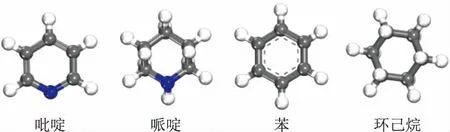
图3 4种分子的结构示意
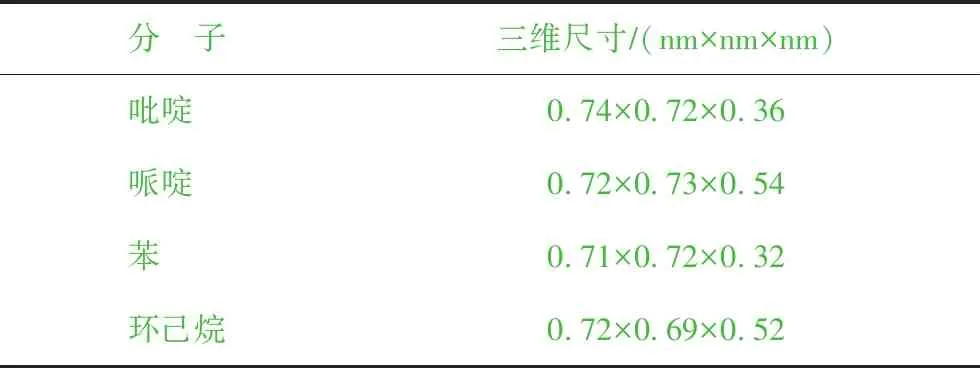
分 子三维尺寸∕(nm×nm×nm)吡啶 0.74×0.72×0.36哌啶 0.72×0.73×0.54苯 0.71×0.72×0.32环己烷0.72×0.69×0.52
1.3 计算参数
使用分子模拟软件Materials Studio 8.0进行计算,其中GCMC模拟使用Sorption模块,应用Metropolis抽样方法,吸附质之间、吸附质与分子筛之间的非键相互作用采用Lennard-Jones势能和Coulomb作用描述。
(1)
式中:ULJ为相互作用能;i和j表示不同原子;Rij表示原子间距,Å(1 Å=0.1 nm);Dij和(R0)ij为Lennard-Jones参数;qi和qj表示原子所带电荷,e。模拟中采用Compass力场,静电作用采用Ewald方法处理,非键相互作用采用Atom based算法,非键作用截断距离设置为1.251 nm,正好小于晶胞边长(2.503 nm)的一半。计算平衡步数为1×106步,生产步数为1×107步。
DFT计算采用Dmol3模块,全局精度控制为Fine,采用GGA-PBE泛函下的DNP基组,对所有原子采用全电子处理;k-point设置为(2×2×1);计算过程中能量、力和位移的精度分别设为1×10-5Ha,0.002 HaÅ,0.000 5 nm。模拟过程中吸附能的计算方法如下:
ΔEads=E-(EM+ES)
(2)
式中:ΔEads表示吸附能,kJmol;E表示客体分子吸附在团簇上时整体的能量,kJmol;EM表示客体分子的能量,kJmol;ES表示团簇的能量,kJmol。
2 结果与讨论
2.1 吸附等温线
在温度为298 K、压力为0~100 kPa的条件下,吡啶、哌啶、苯和环己烷在H-FAU分子筛上的吸附等温线见图4。由图4可知,4种分子的吸附等温线均为 Ⅰ 型等温线。低压下吸附等温线的斜率可用于定性描述主客体分子相互作用的强弱,斜率越大则相互作用越强[19]。从图4可以看出,吡啶和哌啶在低压下的吸附等温线斜率较大,而苯和环己烷的相对较小,表明氮化物与H-FAU分子筛有较强的相互作用。另外,在4×10-3kPa下哌啶的吸附量为3.2 mmolg,此时吡啶仅有0.3 mmolg的吸附量,而苯和环己烷尚未开始吸附,这意味着哌啶吸附的强度更大。
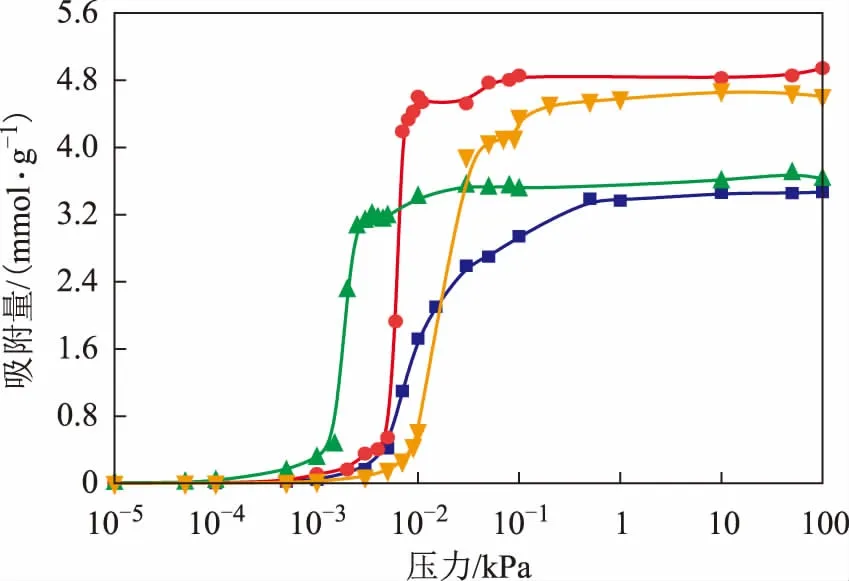
图4 298 K下4种分子在H-FAU分子筛上的吸附等温线●—吡啶; 苯; ▲—哌啶; ■—环己烷
表2汇总了4种客体分子在H-FAU分子筛上的饱和吸附量,吸附量由高到低的顺序为:吡啶>苯>哌啶>环己烷,且吡啶的吸附量是哌啶的1.38倍,是苯的1.03倍,是环己烷的1.47倍,这表明决定吸附量的因素除了相互作用强弱外还有分子的空间位阻效应。对于这4种分子而言,氮化物由于吸附能力强,因此低压下(0~0.1 kPa)吸附速率快,吸附饱和效率高。但由于哌啶的最小截面尺寸为0.73 nm×0.54 nm,在4种客体分子中空间位阻最大,因此单位空间能容纳的哌啶数目小于苯。而吡啶本身与分子筛的相互作用较强,另外其最小截面尺寸与苯接近且小于环己烷,使得吡啶的饱和吸附量最高。
从表2还可以看出,吡啶和苯的吸附量接近,哌啶和环己烷的吸附量接近,但均呈现氮化物的吸附量更高的规律。这表明N取代促进了环状烃在H-FAU分子筛孔道内的吸附。
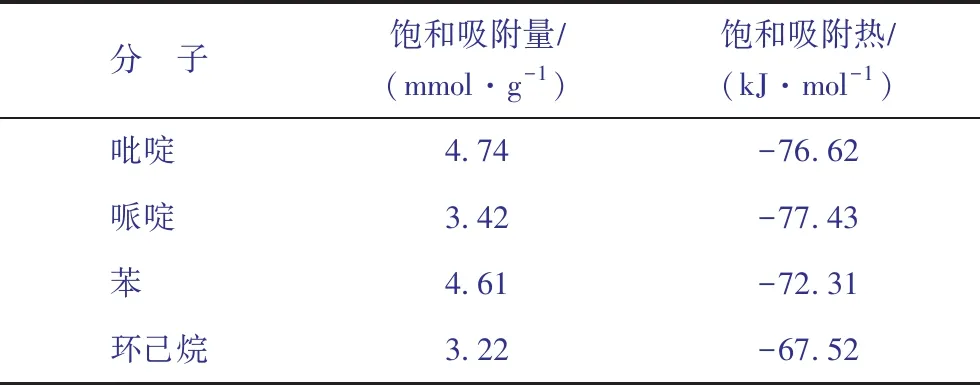
表2 298 K下4种分子在H-FAU分子筛上的饱和吸附量和吸附热
2.2 吸附热
气体分子在多孔材料中吸附时,吸附热是一个重要性质。在GCMC模拟中等量吸附热(ΔE)可由巨正则系综中能量粒子的涨落[20]计算得到,吸附热包含了客体分子间、客体分子和吸附剂之间的相互作用,其大小可用于定量描述吸附强弱。
(3)
式中:T为体系温度,K;R为理想气体常数;N为客体分子数;UN为吸附相势能,kJmol;〈〉为系综平均值。298 K下4种客体分子在H-FAU分子筛上的吸附热曲线如图5所示。从图5可以看出,4种分子的吸附热均随压力的升高(即吸附量的增加)而增大,表明在低压下(0~0.1 kPa)4种气体分子与孔道的相互作用占主导地位,随着吸附量增加,客体分子间的作用增强[21]。从吸附热数值上看,在整个压力范围内吡啶和哌啶的吸附热均大于苯和环己烷,从表2中4种客体分子的饱和吸附热数据可以看出,其由大到小的顺序为:哌啶>吡啶>苯>环己烷,表明氮化物与分子筛之间的相互作用更强,并且碱性更强的哌啶在H-FAU分子筛上的吸附强度大于吡啶,这与2.1节得到的结论一致。
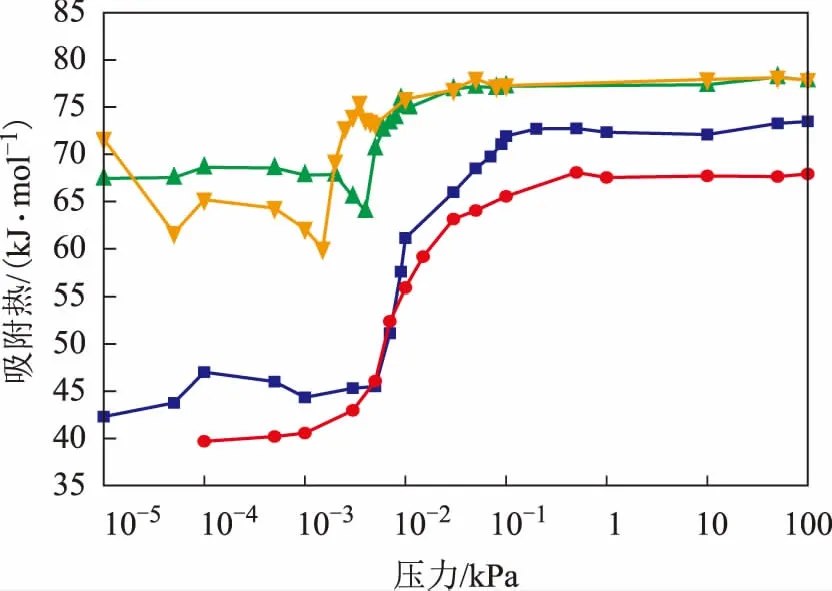
图5 298 K下4种分子在H-FAU分子筛上的吸附热曲线哌啶; ▲—吡啶; ■—苯; ●—环己烷
2.3 吸附质分布
由GCMC模拟可以得到一系列客体分子在吸附剂上的吸附构型数据,将这些数据进行统计分析可以得到它们在多孔材料各个吸附位点出现的概率[19],吸附质分布数据有助于更直观地看清不同客体分子在H-FAU分子筛中的分布情况。
图6为4种气体分子在H-FAU分子筛中的分布情况,红色的区域表示分子可能出现的位置,颜色越深表示出现的概率越大。从图6可以看出,4种分子在H-FAU分子筛的超笼和SOD笼中均有分布,且主要分布在超笼中,而且越靠近超笼中心的区域4种分子分布的密集程度越大。从图6左图中还发现,吡啶和哌啶在分子筛中的分布更集中在表面H质子附近,而苯和环己烷主要离域分布在孔道中,表明H-FAU分子筛上活性位点对吡啶和哌啶的捕捉效率较高。

图6 298 K下4种分子在H-FAU分子筛上的密度分布
综合上述研究表明,吡啶和哌啶与H-FAU分子筛上的H质子有较强的作用,由此推测二者之间可能存在电荷的传递。由于GCMC模拟无法获得电子性质等信息,因此以下应用DFT方法探究4种客体分子与H-FAU分子筛活性中心之间的相互作用。
2.4 客体分子在团簇上的DFT模拟
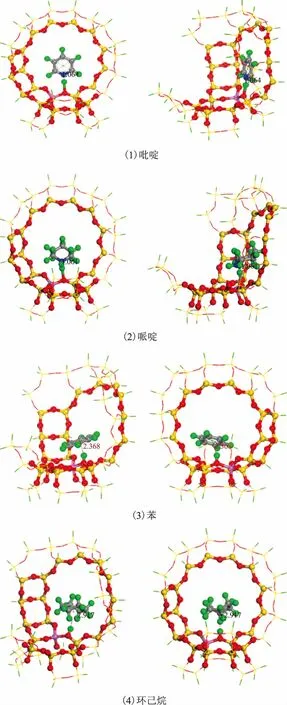
图7 4种分子在H-FAU团簇上的最稳定吸附构型
图7为吡啶、哌啶、苯和环己烷在40T团簇上的最稳定吸附构型,其吸附能和吸附质与活性位点的距离列于表3。苯和环己烷的最稳定吸附构型是以分子的环区朝向H质子,吸附能分别为-84.81 kJmol和-59.27 kJmol。吡啶和哌啶则是以分子上的N原子朝向H质子,而且两者之间形成了化学键,吸附能分别为-201.55 kJmol和-225.77 kJmol。这说明氮化物与FCC催化剂活性中心之间发生强化学吸附,而苯和环己烷主要是物理吸附。从吸附能上看,哌啶的吸附作用最强,因此碱性较强的哌啶对FCC催化剂的毒害作用更大。这是由于吡啶分子中含有共轭双键,相间的π键形成π-π共轭,导致吡啶上的电子更加稳定,与哌啶相比较难提供孤对电子。
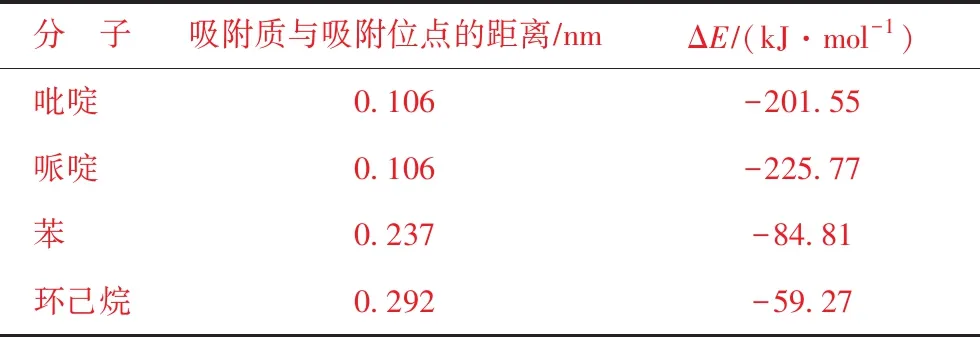
表3 4种分子的吸附数据
分子筛吸附客体分子后结构内总的电子密度不仅可以描述分子筛骨架与客体分子间的相互作用强度,还能够展示电子的分布和转移情况[22-23]。图8为吡啶、哌啶、苯和环己烷在H-FAU团簇上的电子密度分布,在此截取了活性位点和客体分子中心所在的平面,图中红色区域表示高电子密度区,蓝色区域表示低电子密度区。从图8可以看出,苯和环己烷的电子云与H质子的电子云相对孤立,表明二者之间没有发生键合作用,因此吸附能相对较低。另外,由于苯分子两侧具有密度较高的离域π电子,分子与团簇之间的静电作用较强,这导致苯的吸附能大于环己烷。对于吡啶和哌啶,它们的电子云与H质子连成一片,表明这两种氮化物与H质子发生了较强的键合作用。值得注意的是吡啶分子与苯类似,其周围也存在一层离域π电子,π-π共轭的稳定作用导致吡啶的吸附能略低于哌啶。这一结论与Corma等[4]的研究结果一致,他们认为吡啶的中毒效应是由于吡啶吸附后导致H质子电荷密度显著降低,使其无法催化正碳离子反应。上述DFT计算结果从微观角度验证了Corma等的观点。
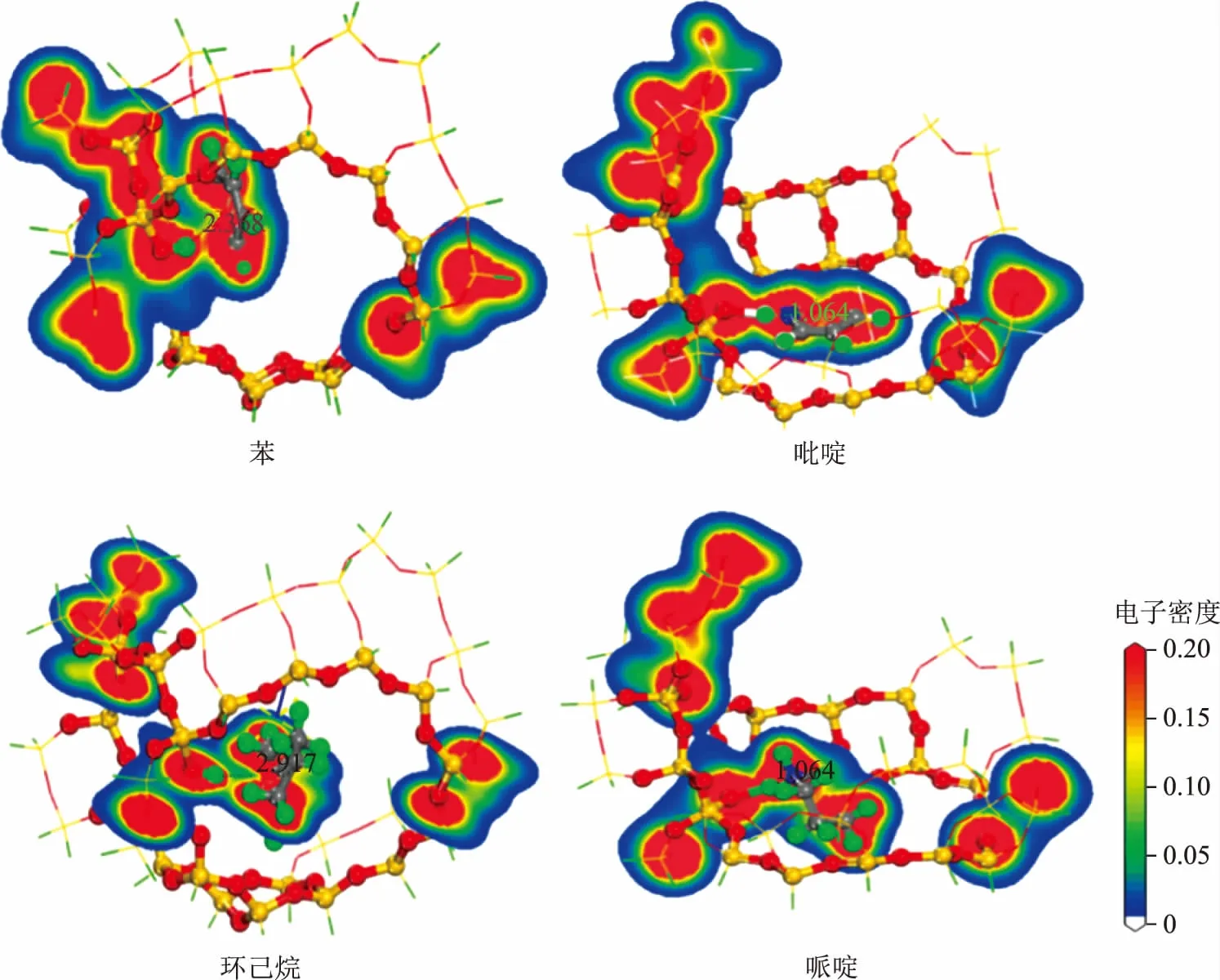
图8 4种分子在H-FAU团簇上的电子密度分布
3 结 论
采用GCMC和DFT方法模拟了吡啶、哌啶、苯和环己烷在H-FAU分子筛上的吸附性质。GCMC计算结果表明,4种分子在分子筛上的吸附量由大到小的顺序为:吡啶>苯>哌啶>环己烷,而吸附热由高到低的顺序为:哌啶>吡啶>苯>环己烷。4种分子主要分布在H-FAU分子筛超笼中,并且吡啶和哌啶在超笼中的分布更加集中,而苯和环己烷主要离域分布在孔道中。DFT计算结果表明,4种分子的吸附能顺序和吸附热相同,苯和环己烷主要以芳环和环烷环朝向H质子,而吡啶和哌啶则是通过N原子与H质子键合,吸附作用更强,电子密度分布也证实了这一结论。模拟结果解释了N取代促进环状烃在分子筛内吸附的原因。
