AMPK/mTOR信号通路的研究进展
2019-09-16张新颖毛景东杨晓燕李树森杜立银
张新颖, 毛景东, 杨晓燕, 李树森, 杜立银
(内蒙古民族大学 动物科学技术学院,内蒙古 通辽 028000)
维持能量稳态和在低营养期间执行适应性反应,是所有细胞的关键功能。在真核细胞中,细胞能量状态的主要传感器是AMP活化蛋白激酶(AMPK)。AMPK是高度保守的新陈代谢调节因子,能在细胞和生理水平的代谢应激下维持能量平衡。细胞不断适应自身的新陈代谢以满足能量需求,并对营养物质的可用性做出反应。一般来说,AMPK通过感知AMP∶ATP和ADP∶ATP的比率变化而被激活,通过抑制消耗ATP的合成代谢过程,促进产生ATP的分解代谢过程,从而恢复能量平衡。此外,AMPK的活性被多个上游信号调节,从而使AMPK成为细胞利用的中心节点,以协调其代谢与特定能量需求[1]。除此之外,AMPK的治疗潜力已被广泛认可,并且用于治疗代谢性疾病如糖尿病、肥胖、炎症和癌症等。mTOR 是生长因子和营养信号的整合器,整合上游的多种信号进而调节细胞的生长。mTORC1的主要功能是调节蛋白质合成和细胞周期进程,而 mTORC2可在肌动蛋白细胞骨架组织和细胞存活方面发挥重要作用。因此,本文主要讨论mTORC1的相关作用机制。AMPK通过对mTOR生长的调节因子作用来调节这些过程。此外,研究表明中性粒细胞是感染、炎症和组织损伤的第一道防线,其依靠趋化性定位感染和炎症部位,mTOR和AMPK参与了中性粒细胞趋化性的调节。细胞生长和蛋白质翻译是细胞中ATP消耗的主要原因。AMPK通过抑制mTORC1 活性降低细胞内的ATP 需求来维持细胞的能量平衡, 一旦能量平衡没有修复, 就会导致细胞自噬的发生。
1 AMP活化蛋白激酶
1.1 AMPK的亚基组成
AMPK是由一个催化亚基α和两个调节亚基β和γ组成的异源三聚体复合物。脊椎动物具有由不同基因编码的每个亚基的多个同工型。在人类中,有两个α亚基(α1和α2),由基因PRKA1和PRKAA2编码;两个β亚基(β1和β2),由PRKAB1和PRKAB编码;三个γ亚基(γ1、γ2 和γ3),分别由PRKAG1、PRKAG2和PRKAG3编码。每个AMPK复合物由一个α亚基、一个β亚基和一个γ亚基组成,并且所有组合都是可能的,因此可能产生12个不同的AMPK复合物[2]。
αβγ复合物有12种可能的组合,取决于哪些亚基构成复合物。在体内,一些细胞类型仅包含这些组合的一个子集,这表明一些复合物具有特定的作用。例如,在肌肉中仅检测α1β2γ1、α2β2γ1和α2β2γ3复合物。此外,运动后只有α2β2γ3复合物被Thr172磷酸化激活。对于含有γ3复合物的这种特异性与线粒体抑制剂PT1的作用形成对比,线粒体抑制剂PT1可以特异性激活肌肉中含γ1的复合物[3]。尽管特定复合物激活的后果尚不清楚,但是这一观察结果表明特定的亚基组成允许不同的AMPK复合物对不同类型的应激刺激作出反应。近年来研究表明,不同的复合物对磷酸酶的活性、核苷酸的活化、磷酸酶的敏感性以及AMP对磷酸酶敏感性的变构调节都有影响[4]。
1.2 溶酶体中的AMPK
最近的研究结果显示AMPK通过与轴蛋白的相互作用而被定位于溶酶体中,轴蛋白最重要的特征是其在WNT通路调节中的作用[5]。在这种情况下,轴蛋白定位于溶酶体,在溶酶体中与肝激酶B1(LKB1,也称为STK11)相互作用。在能量应激下,AMP刺激与轴蛋白的相互作用,使Thr172被LKB1和AMPK激活。这是为了允许在饥饿条件下调节细胞生长的主调节器mTOR和AMPK[6]。 mTOR与AMPK起着相反作用:mTOR在高营养条件下刺激合成代谢途径。AMPK和mTOR都是保守通路的组成部分,控制着分解代谢和合成代谢。研究表明,这种由轴蛋白调节的AMPK可能在葡萄糖饥饿时激活AMPK后起重要作用[7]。此外,葡萄糖饥饿激活的AMPK独立于经典的AMP依赖性变构机制。相反,糖酵解酶醛缩酶与其底物果糖1,6二磷酸(FBP)的结合调节AMPK-轴蛋白的结合和AMPK活性[7]。因此,在葡萄糖饥饿条件下,FBP水平下降,由醛糖酶感知并刺激溶酶体中AMPK-轴蛋白复合物的形成。
1.3 AMPK的活化
AMPK复合物通过上游激酶催化α亚基上的Thr172磷酸化而被激活。AMPK的上游激酶之一是肿瘤抑制因子LKB1,这一发现提供了癌症和代谢之间的直接联系。LKB1(也称为Stk11)基因失活的小鼠的研究表明,在大多数哺乳动物组织中,LKB1是能量应激下大部分AMPK活化的原因,包括肝脏和肌肉等关键代谢组织[8-10]。对于缺乏LKB1细胞的研究表明,LKB1几乎介导AMPK的所有活性,以响应线粒体损伤和低能量条件。然而,AMPK也可以通过钙敏感性激酶CAMKK2(也称为CAMKKβ)响应钙通量而在Thr172上直接磷酸化,从而将钙信号传导与AMPK的能量代谢调节联系起来。细胞内钙的增加可以激活CAMKK2,并导致AMPK的活化,而与LKB1和核苷酸水平无关。研究表明,CAMKK2可以在细胞应激后激活AMPK,包括氨基酸饥饿,缺氧和细胞脱离基质[11-13]。在LKB1缺乏细胞中,CAMKK2仍然能够保持一些对ATP/AMP比率变化敏感的AMPK活性。各种生理条件会导致体内AMPK的活化,包括线粒体抑制、营养不足和运动等。这些条件很可能通过ATP/AMP比例的调节诱导AMPK活化。此外,已发现了几种AMPK的小分子催化剂,这些小分子催化剂与α和β亚基结合,并独立于核苷酸水平激活AMPK激酶活性。这些化合物中的第一种是A-769662[14]。 随后,又发现了具有更高效力和特异性的其他相关化合物,例如化合物991、PF739和MK8722[15-17]。
2 哺乳动物雷帕霉素靶蛋白
哺乳动物雷帕霉素(mTOR)靶蛋白是进化上保守的丝氨酸/苏氨酸蛋白激酶,其调节多种细胞过程,例如细胞生长、细胞周期、细胞存活和自噬。mTOR形成两个功能性复合物,mTORC1作为多蛋白复合物存在,含有mTOR、Raptor、mLST8(GβL)和PRAS40,而mTORC2由mTOR、Rictor、mSin1(MAPKAP1)、Protor(PRR5)和mLST8组成。从酵母到哺乳动物,每个复合物的结构都是保守的。mTORC1直接受细胞能量和营养状态的调节,而mTORC2则不受细胞活力的影响。mTORC1在翻译和自噬的调节中起重要作用,对雷帕霉素的抑制作用敏感。
2.1 mTORC1的上游基因
mTORC1 是 mTOR 两个复合物中较有特点的一个,这个分支通路的一个明显特征是它可感受许多上游信号。mTORC1 通路可汇聚和整合来自生长因子、环境应激、能量、氧化和氨基酸五个方面对细胞的刺激信号来调控蛋白和脂肪合成及自噬等许多重要生理过程。结节性硬化复合物1和2(TSC1/2,tuberous sclerosis 1and2)异二聚体复合物是 mTORC1 上游的重要负性调控因子,对Ras同系物(Rheb,the Ras homolog enriched in brain)GTP酶具有GAP活性。有活性的GTP结合的 Rheb 可直接与mTORC1 相互作用,极大地促进了其激酶活性。TSC1/2 通过将有活性的 GTP 结合的Rheb 转变成无活性的GDP结合形式,从而负性调控mTORC1。
与生长因子信号输入mTORC1的机制一样,许多应激也通过(或部分通过)TSC1/2产生作用,其中以低能量﹑低氧水平和DNA 损坏最为典型。在低氧或低能量状态下,AMPK使TSC2磷酸化并增加了其对Rheb的GAP活性。低氧诱导REDD1(regulation of DNA damage response1)的表达,以一种难以理解的方式激活TSC2的功能。DNA 损坏也通过多个机制给mTORC1 传递信号,这些机制都需依赖p53的转录。DNA损坏诱导TSC2和Pten的表达进而导致整个PI3K-mTORC1信号轴的下调,同时通过依靠对 Sestrin1/2诱导的机制激活AMPK[18]。
2.2 mTORC1的下游基因
mTORC1最典型的底物是核糖体蛋白(RP)S6激酶(S6K)和真核起始因子4E结合蛋白1(4EBP1)(图1)。mTORC1主要磷酸化S6K1的Thr389。S6K1上的Thr389磷酸化募集磷酸肌醇依赖性激酶-1(PDK1),并增强S6K1激活环中PDK1依赖性Thr229磷酸化,这是S6K1激活所必需的过程。S6K1磷酸化许多底物如eIF4B、PDCD4、Skar和S6,从而调节翻译起始,mRNA加工和细胞生长。果蝇S6K1的切除会导致细胞大小和体型的减少。此外,小鼠中S6K1基因的破坏显示出具有小体型的表现。
4EBP1是4EBP家族(4EBP1,2和3)的成员,其作为翻译起始的抑制因子。4EBPs具有真核转录起始因子4E(eIF4E)结合域,由eIF4G共享,eIF4G是形成eIF4F复合物的必需支架蛋白。低磷酸化的4EBP1与eIF4E强烈相互作用,从而干扰eIF4E和eIF4G之间的结合。通过mTORC1依赖的4EBP1磷酸化,4EBP1从eIF4E解离,从而减轻4EBP1对eIF4E依赖性翻译起始的抑制作用。最近的研究表明,4EBPs是调节细胞数量和增殖的mTORC1途径的重要元素。mTORC1还能磷酸化ULK1,这是一种与ATG1同源的哺乳动物激酶,ATG1是一种丝氨酸/苏氨酸蛋白激酶,可引发自噬起始[19]。mTORC1通过对ULK1的磷酸化抑制 ULK1的激酶活性,从而抑制自噬的启动[20]。
3 AMPK通过多种机制抑制mTORC1
大量研究表明,mTORC1活性通过多种机制受到细胞内能量水平的调节。2001年报道了第一个证明细胞ATP水平与mTORC1活性之间关系的证据。2002年证明了AMPK和mTORC1激活之间的相互关系。2003年直接证明了AMPK抑制mTORC1活性的证据。之后,几个研究小组发现AMPK直接磷酸化mTORC1通路中的多种成分(图1)。AMPK磷酸化TSC2并激活TSC,从而衰减TORC1途径。与此模型一致的是,在TSC1或TSC2缺陷细胞[21]中,氨基咪唑羧甲酰胺核苷酸(AICAR)、AMPK激活因子或葡萄糖剥夺对mTORC1活性的抑制作用很大。在AMPKα1α2双敲除MEF(小鼠胚胎成纤维细胞)中,AICAR不能抑制mTORC1活性[22]。 此外,葡萄糖饥饿抑制野生型细胞中的细胞生长,但不抑制TSC1缺陷细胞中的细胞生长。与此同时,缺乏TSC1的细胞,而非野生型细胞,在葡萄糖缺乏的条件下会大量死亡,而雷帕霉素治疗可以防止葡萄糖缺乏的细胞在葡萄糖缺乏的情况下死亡[23-24]。
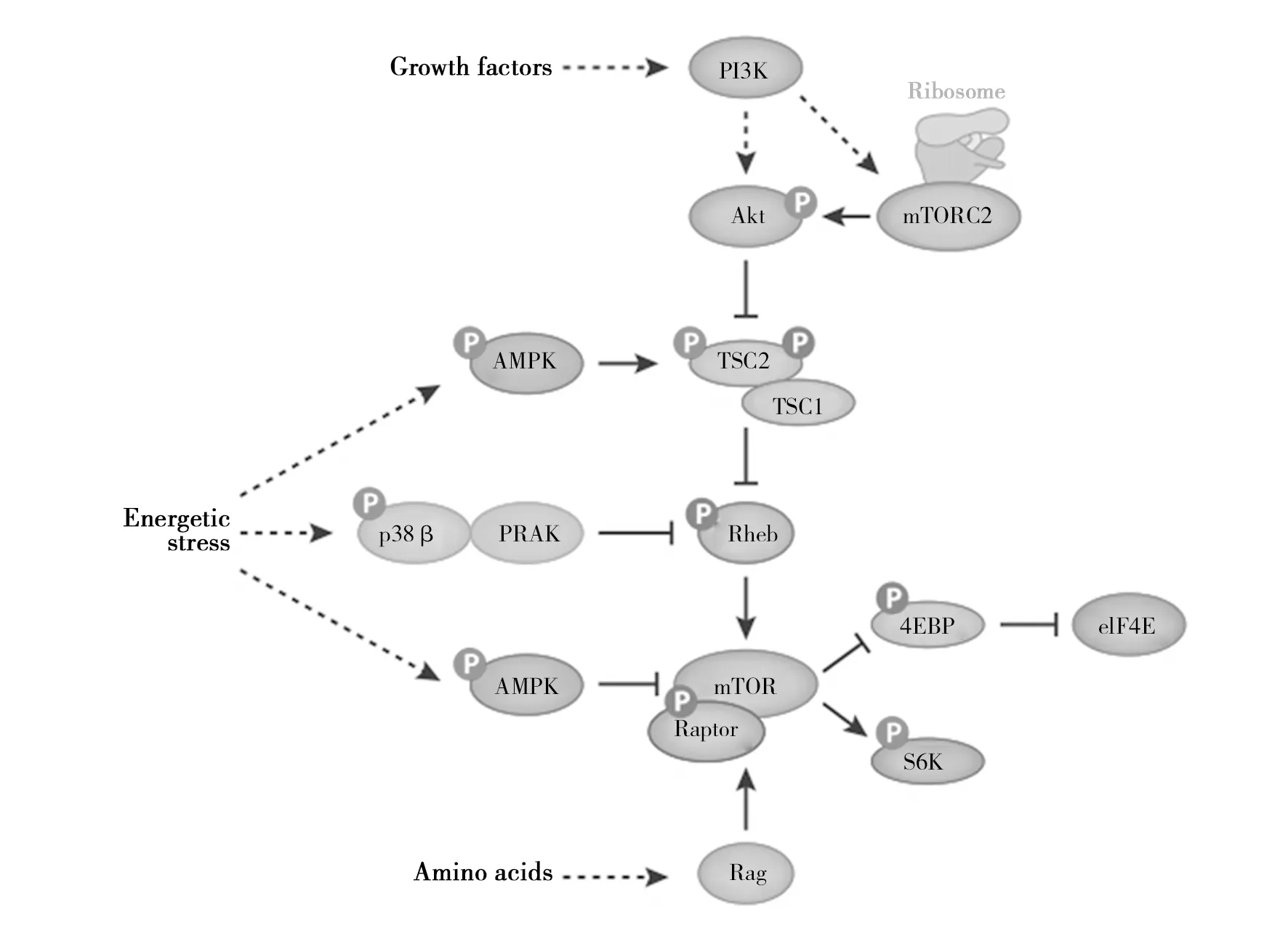
图1 AMPK与mTOR的作用机制Fig.1 Mechanism of action of AMPK and mTORAMP激活蛋白激酶(AMPK)以多种方式抑制哺乳动物雷帕霉素复合物1(mTORC1)的途径。 在能量应激条件下,AMPK磷酸化TSC2(结节硬化复合物)和Raptor以抑制mTORC1通路。来自AMPK and mTOR in Cellular Energy Homeostasis and Drug Targets(Ken Inoki,et al. 2012)AMP-activated protein kinase (AMPK) inhibits the pathway of the mammalian target of rapamycin complex 1 (mTORC1) in multiple fashions. Under energetic stress conditions, AMPK phosphorylates TSC2 and Raptor to inhibit the mTORC1 pathway.From AMPK and mTOR in Cellular Energy Homeostasis and Drug Targets(Ken Inoki,et al. 2012)
在葡萄糖饥饿的情况下,使TSC缺陷细胞发生细胞死亡的机制包括p53肿瘤抑制因子的积累,内质网(ER)应激的增强和存活激酶的减少[23,25]。因此,多种因素可能导致TSC突变细胞对能量饥饿的超敏反应。AMPK是能够通过Ser15磷酸化来稳定p53,p53是一种主要的促凋亡蛋白,mTORC1的组成性激活增强了p53的翻译[23]。综合起来,这两种效应导致大量p53在葡萄糖缺乏的TSC细胞中积累,从而增加细胞死亡易感性。此外,活化的mTORC1信号传导诱导ER应激并激活未折叠的蛋白质反应。由mTORC1激活引起的连续ER应激导致胰岛素敏感途径以及存活通路的减少。活性S6K还导致胰岛素受体底物的下调,并产生对PI3K-Akt途径的负反馈抑制[26-27]。在缺乏TSC细胞中生存信号通路减弱。Blenis及其同事证明了雷帕霉素对TSC缺陷细胞中mTORC1的抑制可以使细胞维持ATP水平,并在葡萄糖缺乏的条件下减弱AMPK的激活[28]。这些数据表明,TSC-mTORC1途径在AMPK的上游起作用。在高能应激条件下,抑制mTORC1对细胞存活能量是至关重要的。综上所述,完整AMPK-TSC信号传导的反应对于抑制mTORC1通路在能量压力条件下调控细胞存活和生长是必需的。
虽然上述机制是在应激条件下调节mTORC1的线性信号通路,但细胞也可以通过与Raptor相关的mTORC1直接调控mTORC1(图1)。在秀丽隐杆线虫和小脑球虫中,mTOR和AMPK的逆调节作用也被保留下来,这两种动物都缺乏TSC2的同源物种,这就增加了在这些有机体中依赖AMPK的mTORC1存在替代调节机制的可能性。Shaw等发现[29],AMPK也使Raptor上的Ser722和Ser792磷酸化。该研究表明,在Ser722和Ser792中表达一种具有磷酸缺陷的Raptor突变体的细胞,可以抵抗AICAR诱导的mTORC1抑制,表明AMPK诱导的Raptor磷酸化对mTORC1活性有负调控作用。然而,最近的一项研究[21]显示,尽管Raptor被AMPK完全磷酸化,AICAR也不能抑制TSC缺陷型成纤维细胞中的mTORC1活性。随着Raptor磷酸化的增加,AICAR抑制TSC缺陷型肝细胞中的mTORC1活性。该研究[21]进一步表明,TSC参与AMPK诱导的mTORC1调控取决于所研究的细胞类型和组织。
最近研究显示,缺糖损伤和AMPK活化因子如AICAR和2-脱氧-葡萄糖(2DG)是抑制mTORC1信号传导的第三种机制。相关学者研究通过磷酸化依赖性机制将p38β-PRAK途径与Rheb活性的下调联系起来(图1)[30]。在葡萄糖饥饿时,AICAR或2DG处理后,p38β-PRAK途径以不依赖于AMPK的方式被激活,并且活化的PRAK直接磷酸化Rheb上的Ser130。Rheb的磷酸化不仅导致其GTP结合的损伤,而且还导致了从Rheb中分离出结合的GTP。尽管尚不清楚Rheb的Ser130磷酸化是否诱导了Rheb GTPase的内在活性,还是刺激了依赖TSC2的Rheb GTP水解,但研究显示,包括GDP在内的鸟嘌呤核苷酸结合减少了。因为p38β-PRAK依赖性的Rheb磷酸化发生晚于AICAR处理细胞中mTORC1活性的初始抑制,TSC2或者Raptor介导的mTORC1抑制可能导致能量应激期间mTORC1抑制的急性期,其中p38β-PRAK的依赖性调控负责持续的mTORC1抑制[22]。
许多研究使用不同的方法和化学物质来激活AMPK。例如,葡萄糖消耗或缺氧等通常用作激活AMPK的手段。此外,已经使用了各种化学品,包括AICAR、双胍(如二甲双胍和苯乙双胍)、糖酵解抑制剂(2DG)和代谢性毒物如寡霉素和FCCP,以研究AMPK在调控mTORC1途径中的作用。
4 AMPK/mTOR调节能量代谢
4.1 糖代谢和脂代谢
葡萄糖和脂质是细胞中能量供应和储存的主要来源。AMPK通过促进其分解,抑制其合成和储存来增加ATP水平。AMPK通过磷酸化TBC1D1(TBC结构域家族,成员1)和TXNIP(硫氧还原互作蛋白)促进葡萄糖摄取蛋白质,其通过不同的机制分别控制葡萄糖转运蛋白GLUT4和GLUT1的易位和细胞表面水平[31-32]。AMPK还通过磷酸化PFKFB3(6-磷酸果糖-2-激酶/果糖-2,6-双磷酸酶3)来调节一些组织类型的糖酵解,同时通过抑制GYS(糖原合成酶)的多个异构体来抑制葡萄糖在一些组织中的储存[31]。 AMPK通过ACC1和ACC2的直接磷酸化来控制细胞整体脂质代谢,通过ACC2在线粒体外膜局部产生的丙二酰辅酶A抑制CPT1(肉毒碱棕榈酰转移酶1),抑制脂肪酸合成并同时促进脂肪酸氧化,ACC2在其氨基末端具有线粒体靶向序列。 AMPK还磷酸化并抑制HMGCR(3-羟基-3-甲基-戊二酰辅酶A还原酶),其共同作用于ACC1和ACC2,导致细胞中脂质和固醇合成的预编程。AMPK还通过磷酸化脂肪酶例如HSL(激素敏感性脂肪酶)和ATGL(脂肪细胞-甘油三酯脂肪酶)促进脂质吸收和释放。
AMPK通过磷酸化和核排斥CRTC2(循环- AMP调控转录共激活因子2)和II类HDACs(组蛋白去乙酰酶)抑制糖异生基因的转录诱导、葡萄糖的从头合成过程,这些都是糖异生基因转录所必需的辅助因子。此外,AMPK磷酸化并抑制转录因子,其激活糖酵解和脂源性转录程序,最显著的是SRBEP1(固醇调节元件结合蛋白1),是脂质合成的主转录调节因子,还包括HNF4A(肝细胞核因子-4A)和ChREBP(碳水化合物应答元件结合蛋白)。因此,急性AMPK激活有利于葡萄糖摄取以促进ATP恢复,而持续的AMPK激活重新编程细胞以限制葡萄糖和脂质的合成,并有利于脂肪酸作为能量源的氧化。
4.2 蛋白质代谢
AMPK抑制蛋白质合成的能力是通过直接抑制mTORC1复合物来实现的。mTOR是营养和生长因子信号的中心整合体,其激活许多生物合成途径,特别是蛋白质翻译,并且可以刺激细胞生长。在许多水平上,AMPK和mTORC1在调节细胞代谢中起反作用。 AMPK通过两种机制抑制mTORC1活性,一种是通过mTORC1的负调节因子TSC2的磷酸化和活化[27],另一种是通过mTORC1复合物的亚基[33]Raptor的磷酸化来抑制mTORC1活性。除了抑制mTOR之外,已报道AMPK通过磷酸化和抑制TIF-IA(转录起始因子IA)阻断核糖体RNA合成来限制蛋白质合成[34]。 AMPK还通过磷酸化和激活eEF2K(真核延伸因子2激酶)来抑制蛋白质的延伸[35]。此外,mTORC1也是eEF2K的主要调控因子[36],为AMPK的许多下游靶点提供了一个例子,这些靶点也被mTORC1或S6K1直接磷酸化,以拮抗AMPK磷酸化对其功能的调节。以这种方式,AMPK和mTOR控制合成代谢和分解代谢,通过在细胞内循环,启动和关闭一组有限的主代谢开关。
5 MPK/mTOR调控自噬
自噬是一种将蛋白质、细胞器和其他大分子传递到溶酶体进行降解的细胞过程。这是一种由细胞利用的过程,用于正常运转和针对能量短缺产生营养物。AMPK通过几种机制有效促进自噬。AMPK磷酸化并激活ULK1,触发自噬级联的启动[37-39]。重要的是,mTOR通过直接磷酸化和抑制ULK1[38]强烈地抑制自噬。因此,AMPK不仅通过直接激活ULK1而且通过负调节mTORC1并阻断其对ULK1的抑制作用来促进自噬。因此,ULK1是AMPK和mTOR以相反的方式调节特定代谢过程的另一个节点。AMPK还通过VPS34(液泡蛋白34)复合物的差异调节来刺激自噬的起始[40],这对于自噬体的起始和形成是重要的。据报道AMPK在不含自噬接头蛋白的非自噬复合物中直接磷酸化和抑制VPS34,同时通过直接磷酸化Beclin-1增强自噬复合体中含有Beclin-1的VPS34活性。以这种方式,AMPK可能抑制非必需的囊泡运输,有利于在营养物饥饿期间膜运输进入自噬途径。鉴于AMPK和ULK1都直接磷酸化Beclin-1和Vps34两个不同部位,因此对于不同应激反应的自噬起始时间和空间控制尚有待澄清。此外,AMPK和ULK1也被报道为磷酸化并控制Atg9的定位。Atg9是一种跨膜蛋白,参与早期自噬体的形成。AMPK最近也被证明通过转录机制促进自噬,主要通过调控Tfeb(转录因子EB),Tfeb是溶酶体基因和自噬的主要转录调节因子。尽管目前尚无文献报道AMPK和Tfeb之间存在直接联系,但AMPK可以通过抑制mTORC1激活Tfeb,从而阻断mTOR磷酸化Tfeb并将其移出细胞核的能力[41]。
6 展 望
大量的生物化学、细胞生物学、遗传学和生理学研究证实AMPK是关键的细胞能量传感器,其活化促进能量分解代谢并抑制能量消耗合成代谢。许多AMPK底物是直接参与能量代谢的代谢酶,如糖酵解和脂肪酸合成和氧化。此外,AMPK通过自噬诱导细胞内容物如蛋白质和细胞器的水解,这部分通过抑制mTORC1和激活ULK1来实现。mTORC1整合细胞营养状态,包括能量水平,并在细胞生长中起主要作用。高mTORC1活性促进细胞生长,而低mTORC1活性抑制生长并诱导自噬。在营养不足的情况下,mTOR活性降低导致核糖体生物合成和蛋白质翻译的减少,这通常消耗大量的细胞能量。因此,mTORC1不仅在营养方面起着关键作用,而且在细胞能量稳态中起着关键作用。
AMPK和mTORC1是关键的细胞营养指标和细胞生长调节因子。因此,该通路的失调与许多人类疾病有关。例如,通过TSC1/TSC2中的突变或PI3K通路的组成性活化而不受控制的mTORC1激活会导致肿瘤发生。因此,已经显示mTORC1抑制剂对TSC疾病具有治疗作用。此外,mTORC1抑制剂雷帕霉素类似物已被批准用作治疗晚期肾癌的药物,并且许多临床试验正在使用mTOR抑制剂进行癌症治疗。mTORC1还可以通过自噬调节蛋白质代谢,靶向合成和降解; 因此,mTORC1的抑制可能有助于开发有关蛋白病的药物,包括神经退行性疾病如阿尔茨海默病。同样,AMPK激活的缺陷可能与肿瘤发生有关,因为上游激酶LKB1长期以来被认为是肿瘤抑制因子。据报道二甲双胍是最广泛使用的糖尿病药物,可以抑制癌症,因为使用二甲双胍的患者癌症的发生率显著降低。此外,AMPK和mTOR在能量代谢中的突出作用使其成为糖尿病和肥胖症等代谢性疾病的药物靶点。最后,抑制mTORC1可以延缓秀丽隐杆线虫、果蝇和小鼠的衰老,延长寿命。因此,AMPK和mTORC1都将继续作为突出的药物靶点受到制药行业的广泛关注。
