Formulation of a film-coated dutasteride tablet bioequivalent to a soft gelatin capsule(Avodart®):Effect of γ-cyclodextrin and solubilizers
2019-09-11MiHongMinJinHyongParkMiRanChoiJongHyunHurByungNakAhnDaeDukKim
Mi-Hong Min,Jin-Hyong Park,Mi-Ran Choi,Jong-Hyun Hur,Byung-Nak Ahn,Dae-Duk Kim
aCollege of Pharmacy and Research Institute of Pharmaceutical Sciences,Seoul National University,Seoul 08826,Republic of Korea
bCentral Research Institute,Whanin Pharmaceutical Company,4F,GBSA,107 Gwanggyo-ro,Yeongtong-gu,Suwon 16229,Republic of Korea
Keywords:Dutasteride Tablet Cyclodextrin Complex Formulation
ABSTRACT The aim of this study was to optimize a tablet formulation of dutasteride that is bioequivalent to a commercially available soft gelatin capsule(Avodart®).The effect of cyclodextrin on enhancing the aqueous solubility of dutasteride was investigated,after which the formulation was further optimized with solubilizing polymer and surfactant.Among the cyclodextrins tested, the highest solubility was observed when dutasteride was complexed with γ-cyclodextrin. Moreover, the addition of polyvinylpyrrolidone and Gelucire/TPGS further enhanced the solubility of dutasteride.Differential scanning calorimetry(DSC)and powder X-ray diffraction (pXRD) studies demonstrated that dutasteride existed in the amorphous form in the complex.Optimized dutasteride complexes were selected after a pharmacokinetic study in rats,and film-coated tablets were prepared by the direct compression method.In vitro dissolution profiles for the tablets of dutasteride complexes were similar to those of the reference. Moreover, pharmacokinetic parameters including the Cmax and AUC values after oral administration in beagle dogs were not significantly different from those of the reference with a relative bioavailability of 92.4%.These results suggest the feasibility of developing a tablet formulation of dutasteride using cyclodextrin complex in addition to a solubilizing polymer and surfactant.©2018 Shenyang Pharmaceutical University.Published by Elsevier B.V.This is an open access article under the CC BY-NC-ND license.(http://creativecommons.org/licenses/by-nc-nd/4.0/)
1. Introduction
Dutasteride is a competitive inhibitor of type I and type II 5-α-reductases and is used to treat benign prostatic hyperplasia(BPH)and hair loss[1].Studies have revealed that dutasteride can reduce fetal adrenal and prostate weight and can increase fetal ovarian and testis weight.It has been classified as pregnancy category X by the FDA; thus, women who are pregnant or may become pregnant must avoid taking and handling dutasteride.
Dutasteride is classified as Biopharmaceutics Classification System(BCS)class II and is commercially available in the market only as a soft gelatin capsule formulation due to its low aqueous solubility [1]. However, the physical strength of the gelatin shell could become weaker under high temperature, which might break the seam-line or deform the shape of the capsule. Additionally, the active ingredient could migrate into the gelatin shell [2].Because dutasteride is readily absorbed through the skin, these issues can lead to various health problems. Therefore, developing a tablet form of dutasteride is required to enhance the safety of the drug. Additionally, improved patient compliance is expected with a smaller solid tablet than a soft gelatin capsule.Moreover,because dutasteride is commonly co-prescribed with other BPH medicines such as tamsulosin, it would be more convenient to formulate solid dosage forms for fixed-dose combinations with other drugs.
Previous studies on solubilization of dutasteride have been mainly focused on self-emulsifying drug delivery system(SMEDDS) technology [3-5], which is an oil formulation suitable for soft capsule.To increase the bioavailability of various hydrophobic and poorly water-soluble drugs, the drugs can be formulated to form a complex with cyclodextrin (CD) as a solid dosage form, thereby enhancing their solubility and/or dissolution rate [6-12]. Because no covalent bonds are involved in the drug-CD complex formation,the complex can be easily dissociated in aqueous solution [13].Moreover,diverse approaches have been attempted to further enhance the complexation efficacy,which include the addition of polymers[14],organic salts[15],and buffer[16]to the complexation media.Addition of a small amount of a water-soluble polymer to an aqueous complexation medium increases the complexation efficiency, which consequently can decrease the formulation bulk by reducing the amount of CD required[13].Moreover, water-soluble polymers form complexes with various compounds and stabilize micelles and other types of aggregates in aqueous solutions[13,17].They are additionally capable of increasing the aqueous solubility of cyclodextrins without decreasing their complexing abilities[18].Pharmaceutical polymers such as methylcellulose, hydroxypropylmethylcellulose and polyvinylpyrrolidone have traditionally been used to prevent drug nucleation and crystal growth by creating a polymeric network around growing crystals [19]. Thus, their addition leads to a decrease in drug crystallization and generates a synergetic effect on the solubilizing effect of CDs[8].
Additionally, we assume that the addition of surfactants would further enhance the solubilization of free drug dissociated from the drug-CD complex. The objective of this study was to investigate the effect of the CD complex on enhancing the aqueous solubility and dissolution of dutasteride, after which the formulation was further optimized with diverse polymers and/or surfactants.After a film-coated tablet formulation was finalized,its pharmacokinetics in beagle dogs was compared to that of Avodart®soft capsule.
2. Materials and methods
2.1. Materials
Dutasteride was purchased from Cipla Ltd (Mumbai, India).α-Cyclodextrin (α-CD), β-cyclodextrin (β-CD), γ-cyclodextrin(γ-CD) and hydroxypropyl-β-cyclodextrin (HP-β-CD) were obtained from Wacker Chemie AG (München, Germany).Polyvinylpyrrolidone K30 (PVP) (BASF, Germany), D-αtocopheryl polyethylene glycol 1000 succinate (TPGS)(Isochem,France),stearoyl polyoxylglycerides(Gelucire 50/13)(Gattefosse, France), polyethyleneglycol (PEG400) (Yakuri Pure Chem, Japan) and polyethyleneoxide-polypropylene oxide copolymer (Poloxamer 407) (BASF, Germany) were used as solubilizers. Lactose (SuperTab 11SD) (DFE pharma,Japan), microcrystalline cellulose (Avicel PH102) (FMC, USA),crospovidone (Polyplasone XL) (Ashland, Netherland), magnesium stearate (Faci, Italy), Opadry®(Colorcon, Singapore)and ethylcellulose (Ethocel 10) (Colorcon, Korea) were used as excipients for the tablets. Avodart®soft capsules (Glaxo-SmithKline, United Kingdom) were purchased from a local pharmacy.
2.2. Preparation of dutasteride-cyclodextrin complex and solubility study
Dutasteride-loaded CD complexes were prepared by the ovendrying method. Briefly, dutasteride was first dissolved in ethanol at 2 mg/ml concentration.Various types of cyclodextrins (α-CD, β-CD, γ-CD, HP-β-CD) were separately dissolved in distilled water (DW) at a concentration of 100 mg/ml. The dutasteride solution and CD solution were homogeneously mixed at a 1:1 volume ratio,followed by drying in an oven at 60°C (SANYO, Japan), to determine the aqueous solubility of dutasteride complexed with various CDs at a 1:50 weight ratio.Dried dutasteride-cyclodextrin(DuCD)complexes(equivalent to approximately 0.5 mg of dutasteride)were dispersed in 1.0 ml of DW. After gentle stirring for 1 h, undissolved dutasteride was removed through filtration (0.45-μm PVDF filter),followed by appropriate dilution with a mixture of acetonitrile and water (60/40, v/v). The concentration of dutasteride was analyzed using high-performance liquid chromatography(HPLC),equipped with a reverse phase C18column(Zorbax SBphenyl,150 mm × 3 mm,3.5 um,Agilent) and UV detector at 240 nm. The mobile phase was a mixture of acetonitrile and water (55/45, v/v) at a flow rate of 0.5 ml/min. The injection volume was 50 μl[20].
Because the γ-CD complex exhibited the highest solubility among the complexes tested,complexes were prepared at various weight ratios (1:10-1:70) of dutasteride to γ-CD (Duγ CD)to optimize the solubility of dutasteride. Next, a 0.4 or 1.0 weight ratio of polymer and/or surfactant was added to the dutasteride-γ-cyclodextrin complex (Duγ CD-PS) as a solubility auxiliary additive to further enhance the aqueous solubility of dutasteride.The aqueous solubility of dutasteride in the Duγ CD and Duγ CD-PS solutions was determined after filtration as described above.
2.3. Pharmacokinetics after oral administration of DuγCD-PS complex in rats
The pharmacokinetics of dutasteride after oral administration of diverse Duγ CD-PS complexes was compared with that of the 参考文献(Avodart®,GlaxoSmithKline)in rats.The animal studies were approved by the WhanIn Pharmaceutical Company Animal Ethics Committee. Male Sprague-Dawley rats(8 weeks old, 230-270 g) were purchased from DBL Co., Ltd(Chungcheongbuk-do, Korea). All rats were habituated for 1 week before the experiment and randomly divided into groups of 4-6 animals each. The rats were subjected to fasting 12 h prior to the study, and the carotid arteries were cannulated with polyethylene tubing PE-50 under isoflurane(I-FRAN LIQUID, Hana Pharm Co., Ltd., Seoul, Korea). Each group of animals was administered either the 参考文献 drug (interior oil content of Avodart®soft capsule)or Duγ CD-PS complex(suspended in DW) via oral gavage at a dose of 2.39 mg/kg of dutasteride,and each rat was orally administered 10 ml/kg of DW.Blood samples(approximately 0.3 ml)were collected from the carotid artery into heparinized tubes at 0, 0.5, 1, 2, 4, 8, and 24 h after the administration. The plasma was obtained by centrifuging the samples at 13,000 rpm for 5 min and stored at-70°C until analysis.
The concentration of dutasteride in the plasma samples was analyzed using LC/MS/MS, as previously described [21].Briefly,100 μl of plasma samples was vortex mixed with 900 μl of acetonitrile containing finasteride(10 ng/ml)as an internal standard and centrifuged at 13 000 rpm. Next, 5 μl of supernatant was injected into the LC/MS/MS system.LC separation was performed by an Acquity H class UPLC (Waters, USA),and the mass spectrometric detection was performed on a TQ Detector (Waters, USA) using MRM. A turbo electrospray interface was used in positive ionization mode. The major working parameters of LC and the mass spectrometer are summarized in Supplement Table S1. The pharmacokinetic parameters (Tmax, Cmax, and AUC0-24h) of dutasteride were analyzed using WinNonlin®(ver.6.2,Pharsight) based on the linear trapezoidal rule.The relative bioavailability (BA) of the Duγ CD-PS complexes was calculated as follows:
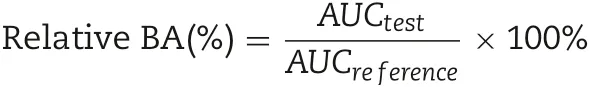
2.4. Characterization of dutasteride and γ-cyclodextrin complexes
The surface morphology was observed using field emission scanning electron microscope (FESEM) (JSM-6700F, JEOL,Japan)at an accelerating voltage of 5 kV.Samples were spread onto carbon tabs (double-adhesive carbon-coated tape) adhered to aluminum stubs,which were then coated with a thin layer of platinum.Thermal analysis of Duγ CD and Duγ CD-PS complexes were conducted by using a differential scanning calorimeter (DSC 200 F3 Mala, Netzsch). Analyses were performed in an aluminum pan under a heating rate of 10°C/min over a temperature range of 20-280°C.XRD Ultima III(Rigaku)was used to perform the powder X-ray diffraction(pXRD)analyses.The measurement conditions were as follows:scanning speed of 3°/min and step width of 0.02°. FTIR was observed using Nicolet IR Spectrometer(iS50,Thermo,USA).
2.5. Preparation of dutasteride tablets
Tablets of Duγ CD-PS complexes(F4 and F5)were prepared by the compression method. Briefly, the Duγ CD-PS complexes were granulated using the fluid-bed granulator(WBF-II,Enger,Taiwan)with a mixture of lactose(Super Tab 11SD)and microcrystalline cellulose(Avicel PH 102)as a powder bed.The formulation was designed for each tablet (240 mg total weight)to contain 0.5 mg of dutasteride.Carr’s index for the granules before tablet compression was 18, indicating fair flowability.The granules were compressed on a rotary tablet compressor using an 8.5-mm round shape punch,and the hardness of the tablet was adjusted between 12 and 13 kp.Next,the tablet was film-coated with HPMC-based Opadry®.The dimension of the film-coated tablet after 3% weight coating (diameter 8.5 mm,thickness 4.2 mm, round shape) was smaller compared with the marketed soft gelatin capsule Avodart®(length 19 mm,thickness 6.7 mm,rod shape).(Fig.S1).
2.6. Dissolution test of the dutasteride tablet
In vitro dissolution profiles of dutasteride from the Duγ CD-PS complex tablet were evaluated by the USP dissolution method(Tier I and Tier II) and compared with that of the reference(Avodart®).In the Tier I method,the dissolution rates of dutasteride were measured using apparatus 2 in which the dissolution medium was 900 ml of 0.1 N HCl solution with 2% (w/v)sodium lauryl sulfate (SLS) at 37°C and stirred at 50 rpm. In the Tier II method,the dissolution medium was 450 ml of 0.1 N HCl solution with pepsin (1.6 g/l,label activity 1:3000) for the first 25 min,followed by the addition of 450 ml of 0.1 N HCl solution with SLS (4%, w/v) for the remaining dissolution test.The samples (5 ml) were obtained at fixed time intervals and were analyzed by HPLC with a UV detector,as described above,after filtering through a 0.45 μm PVDF filter.
2.7. Pharmacokinetic study of the dutasteride tablet in beagle dogs
An in vivo cross-over pharmacokinetic study of dutasteride was performed after oral administration of the Duγ CD-PS complex tablet or the reference (Avodart®) in beagle dogs.The animal studies were approved by the Institutional Animal Care and Use Committee of Korea Animal Medical Science Institute. Six male beagle dogs (10 kg, 10 months old)were subjected to fasting overnight before the experiment.Each dog was administered either one capsule of the reference (Avodart®, 0.5 mg as dutasteride) or one tablet of Duγ CD-PS (F5) (0.5 mg as dutasteride), followed by 10 ml of water.Blood samples were taken from the cephalic vein and collected(3 ml)into heparinized tubes at 0,0.5,1,2,4,8,12,24,and 48 h after the administration. The plasma was obtainedby centrifuging the samples at 3000 rpm for 5 min and stored at -70°C until analysis. The wash-out period between treatments was 4 weeks.The treatment of the plasma samples and the LC/MS/MS analysis conditions were the same as that used above for the rat study.The Cmaxand Tmaxwere determined from the experimental data.The calculated dutasteride concentrations were used to obtain the area under the plasma concentration-time profile from time zero to the last concentration time point (AUC0-t) by the linear trapezoidal method.Statistical analysis was performed with the unpaired t-test where appropriate. Significance was set at P <0.05. All data were presented as mean±standard deviation.

Table 1 - Aqueous solubility of dutasteride complexed with various cyclodextrins at a 1:50 weight ratio and the average binding affinity as obtained by the computer docking simulation tool Glide(Schrödinger,New York,USA).

Table 2 - Effect of the weight ratio of dutasteride:γcyclodextrin (DuγCD) on the aqueous solubility of dutasteride.
3. Results and discussion
3.1. Effect of cyclodextrin complex on the aqueous solubility of dutasteride
Table 1 presents the aqueous solubility of dutasteride when complexed with various cyclodextrins at a 1:50 weight ratio,together with their binding affinity obtained by the computer docking simulation tool Glide (Schrödinger, New York, USA).Among the CDs tested,the γ-CD complex resulted in the highest aqueous solubility of dutasteride and showed the lowest binding affinity value, indicating stable complex formation.Thus, γ-CD complexes with various weight ratios (1:10-1:70)of Duγ CD were prepared, and the aqueous solubility of dutasteride was determined. The aqueous solubility of dutasteride increased up to a 1:70 weight ratio (Table 2), and this value was thus selected for further evaluation. It is interesting to note that the solubility of dutasteride synergistically increased with the addition of a solubilizing polymer at a 0.4 weight ratio (i.e., PVP and PEG) and a surfactant (i.e., Gelucire,TPGS and Poloxamer)to the Duγ CD complex(dutasteride:γ-cyclodextrin=1:70) (Table 3). The highest solubility of dutasteride achieved with the addition of the 0.4 weight ratio of PVP and Gelucire was 147 μg/ml,which is 1.5 times higher than the solubility of Duγ CD(1:70)(93 μg/ml).In a previous report,the highest solubility of dutasteride was only 47.1 μg/ml when dutasteride was complexed with HP-β-CD and HPMC at a weight ratio of 1:26.6:13.3[22].Moreover,the study prepared the complex by the supercritical fluid manufacturing method,which is environmentally friendly but not widely equipped in pharmaceutical companies. Thus, it is notable that Duγ CDPS prepared by the simple drying method achieved an aqueous solubility of dutasteride that was higher than the reported solubility.
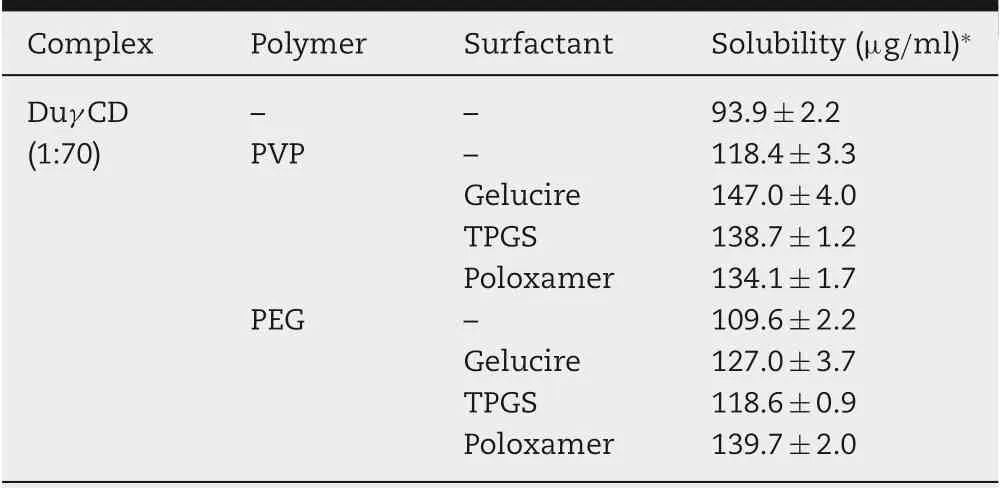
Table 3-Effect of the solubilizing polymer and surfactant on the aqueous solubility of dutasteride(μg/ml)added to the DuγCD (1:70) complex at weight ratios of 0.4 and 1.0,respectively.
Based on the preliminary solubility study,Duγ CD-PS complexes were further optimized by changing the weight ratio of dutasteride to γ-CD (1:10-1:70) and surfactant. Table 4 presents the composition of Duγ CD-PS complexes selected for further evaluation and the aqueous solubility of dutasteride. When the 0.4 weight ratio of PVP was selected as a solubilizing polymer, the solubility of dutasteride increased up to 170 μg/ml as the content of the surfactant(Gelucire:TPGS=1:1) increased to 2 weight ratio (F5).These results are consistent with previous reports that the addition of a water-soluble polymer synergistically enhances the solubilizing effect of CDs by preventing drug nucleation and/or crystal growth [8]. Moreover, it is notable that the surfactant further increased the solubility of dutasteride, which supports our assumption that surfactants would further enhance the drug solubility by solubilizing the free drug dissociated from the drug-CD complex.

Fig.1-Mean plasma concentration-time profiles of dutasteride after oral administration of the DuγCD-PS complex at a dose of 2.39 mg/kg of dutasteride in rats(n=4-6).Each point and vertical bar represent the mean and standard deviation,respectively.
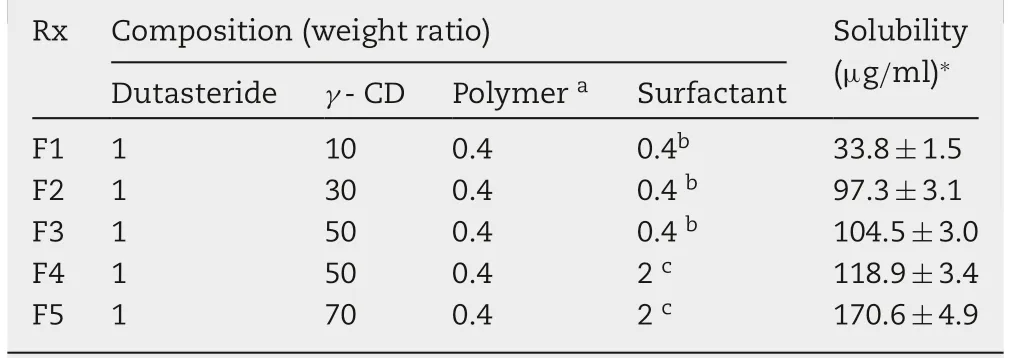
Table 4 - Composition of DuγCD-PS complexes and the aqueous solubility of dutasteride.
3.2. Pharmacokinetic study of DuγCD-PS complex in rats
The plasma concentration profiles of dutasteride after oral administration of Avodart®or the Duγ CD-PS complex at a dose of 2.39 mg/kg of dutasteride in rats are presented in Fig.1,and the pharmacokinetic parameters are summarized in Table 5.Duγ CD-PS complexes(F1)with 10 and 0.4 weight ratios of γ CD and surfactant,respectively,resulted in only 29.6%relative BA compared to that of the reference(Avodart®).However,when the weight ratio of γ CD increased to 30 and 50 (F2 and F3,respectively), the relative BA of dutasteride increased up to 74% of the reference. Moreover, the addition of surfactant at a weight ratio of 2 (F4 and F5) further increased the relative BA up to 93.6%of the reference.
For BCS class II drugs including dutasteride,improving the aqueous solubility is the most practical strategy to increase its oral bioavailability by enhancing the dissolution of the drug in the gastrointestinal (GI) tract [23]. As the solubility of dutasteride increased by increasing the drug to γ-CD weight ratio and by adding surfactant (Table 4), the relative BA of dutasteride increased proportionally(Table 5).In addition to the solubilizing effect of γ-CD, surfactant appears to synergistically enhance the bioavailability (F3 vs.F4) by inhibiting precipitation and solubilizing the free drug dissociated from the CD complex,as we assumed.However,it is interesting to note that the increase in the γ-CD weight ratio up to 70 (F5) could not further increase the relative BA of dutasteride,despite its higher solubility than F4 formulation. It is known that γ-CD solubilizes poorly water-soluble drugs by inclusion of insoluble drug into its hydrophobic cavity and formation of γ-CD aggregates [15]. However, the drug could additionally be embedded among γ-CD aggregates at a high CD ratio,resulting in supersaturation of the drug.The drug is easily released by the dissociation of γ-CD aggregates (by dilution in the gastrointestinal fluid and/or by the ring opening of γ-CD with the attack of digestive enzyme),after which the drug may be precipitated.Surfactant might not be able to sufficiently inhibit the precipitation of supersaturated dutasteride in F5 in vivo;thus,the higher solubility of dutasteride compared to F4 could not proportionally increase the bioavailability.
3.3. Characterization of dutasteride and γ-cyclodextrin complexes
Surface morphology of dutasteride observed by the FESEM was irregular in shape,while γ CD was a parallelogram shape.However, the morphology of complexes was similar to an aggregate (Fig. S2). In FTIR study, characteristic peaks of dutasteride in the range of 1650-1700 cm-1(carbonyl stretching),1600 cm-1(N-H bend)were markedly decreased in γ-CD complexes (Fig. S3). DSC and pXRD are useful techniques todetermine the occurrence of inclusion of drug crystals. The thermograms of dutasteride and the complexes are presented in Fig.2A.Dutasteride showed a very sharp endothermic peak at 251°C,which corresponds to its melting temperature.It is notable that the endothermic peak of dutasteride was almost negligible when the weight ratio of γ CD was higher than 1:30 in the Duγ CD complexes. Moreover, as shown in Fig. 2B, the endothermic peak of dutasteride completely disappeared in Duγ CD-PS complexes (F3, F4 and F5) containing solubilizing polymer (i.e., PVP) and surfactant (i.e., Gelucire and TPGS),indicating that dutasteride is present in an amorphous form in Duγ CD-PS complexes. Moreover, the pXRD results of the Duγ CD-PS complexes were consistent with those of DSC and did not exhibit the specific pattern for dutasteride crystal(Fig. 3). Because both F4 and F5 formulations could achieve high bioavailability (93.6%) compared to the references, they were selected for preparing the tablet formulation.
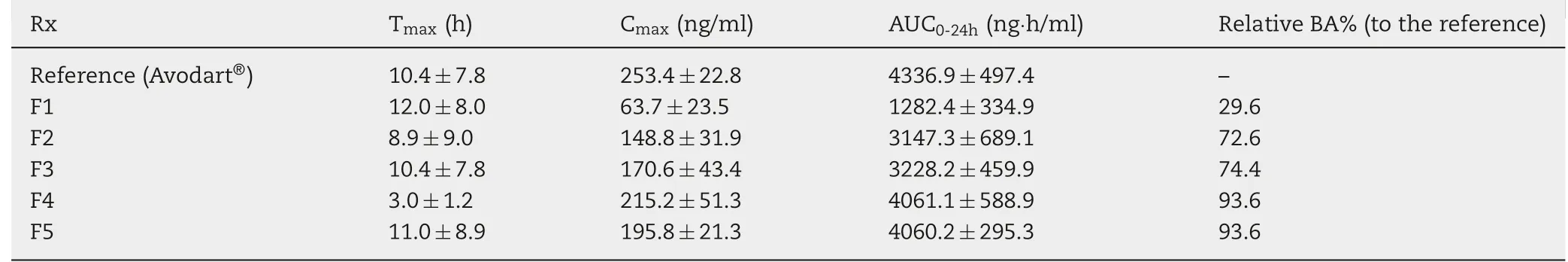
Table 5 - Pharmacokinetic parameters of dutasteride after oral administration of the reference (Avodart®) or DuγCD-PS complex at a dose of 2.39 mg/kg of dutasteride in rats.

Fig.2-DSC thermograms of the(A)DuγCD complexes and(B)DuγCD-PS complexes.
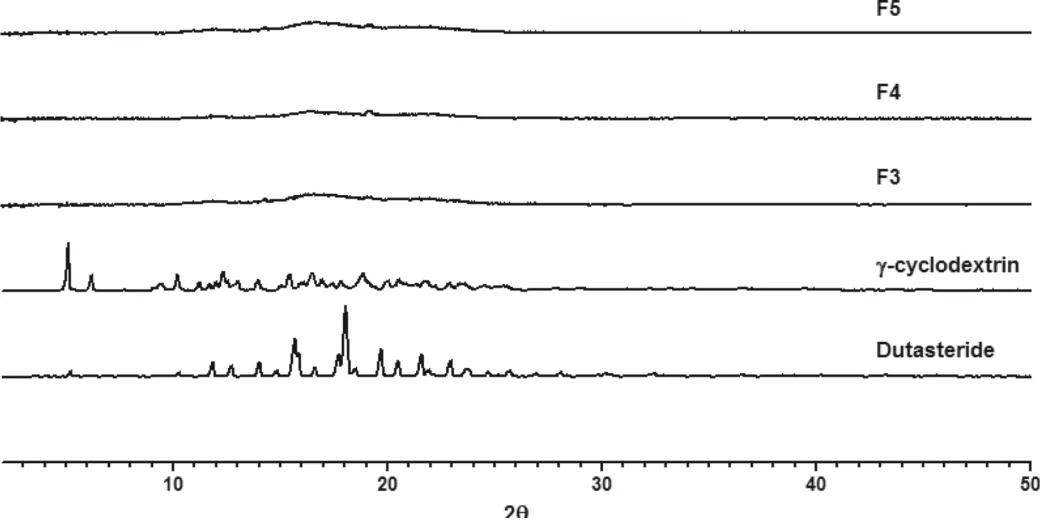
Fig.3-Powder X-ray diffraction pattern of the DuγCD-PS complexes.
3.4. Dissolution study of DuγCD-PS tablet
Fig. 4 presents the in vitro dissolution profiles of dutasteride from the tablets of Duγ CD-PS complexes (F4 and F5) coated with HPMC-based Opadry®, which were compared with that of the reference (Avodart®,soft gelatin capsule).In the Tier I method (Fig. 4A), dissolution of F5 was more rapid than that of F4 and the reference for the first 15 min. However, both F4 and F5 tablets showed a complete release of dutasteride within 45 min,which is similar to the reference.Notably,dutasteride was not dissolved for the first 25 min until SLS was added in the Tier II method(Fig.4B),indicating the importance of surfactants in the release medium to mimic the physiological condition.Moreover,it was previously reported that poorly water-soluble drugs can be solubilized in the gastrointestinal tract by endogenous surfactants including bile acids,bile salts and lecithin[24].

Fig.4-In vitro dissolution profiles of dutasteride from the reference soft gelatin capsule(Avodart®)and the film-coated tablets of DuγCD-PS complexes determined following the USP dissolution method(A)Tier I and(B)Tier II by using apparatus 2.Each point and vertical bar represent the mean and standard deviation,respectively(n=3).

Table 6-Pharmacokinetic parameters of the reference or F5 tablet after oral administration in beagle dogs(n=6,crossover study).
Dutasteride is classified as a BCS class II drug, which implies that it is highly membrane permeable and lipophilic with a log P value of 5.09.Its terminal elimination half-life is known to be very long (3-5 weeks) at steady-state in humans [21].Thus, rapid initial dissolution of dutasteride from the tablet followed by gastrointestinal absorption would be critical to achieve a similar pharmacokinetic profile after oral administration.Based on the in vitro dissolution study(Fig.4A),the F5 tablet was selected for the in vivo animal study.
3.5. Pharmacokinetic study of DuγCD-PS tablet in beagle dogs
Fig.5 shows the plasma concentration profiles of dutasteride after oral administration of Avodart®or the tablet of Duγ CDPS complexes (F5) in beagle dogs.Their pharmacokinetic parameters summarized in Table 6 indicate that the Cmaxand AUCtvalues were not significantly different(P >0.05).Notably,the Tmaxvalue of F5 was shorter than that of the reference,which is consistent with the in vitro dissolution study (Fig.4).Moreover,the relative bioavailability of F5 was 92.4%of that of the reference.Thus,further investigation would be necessary with a larger number of animals and/or humans to evaluate the bioequivalence of the F5 tablet to the reference soft capsule,considering the highly variable plasma concentration of dutasteride(CV=47%)[25].
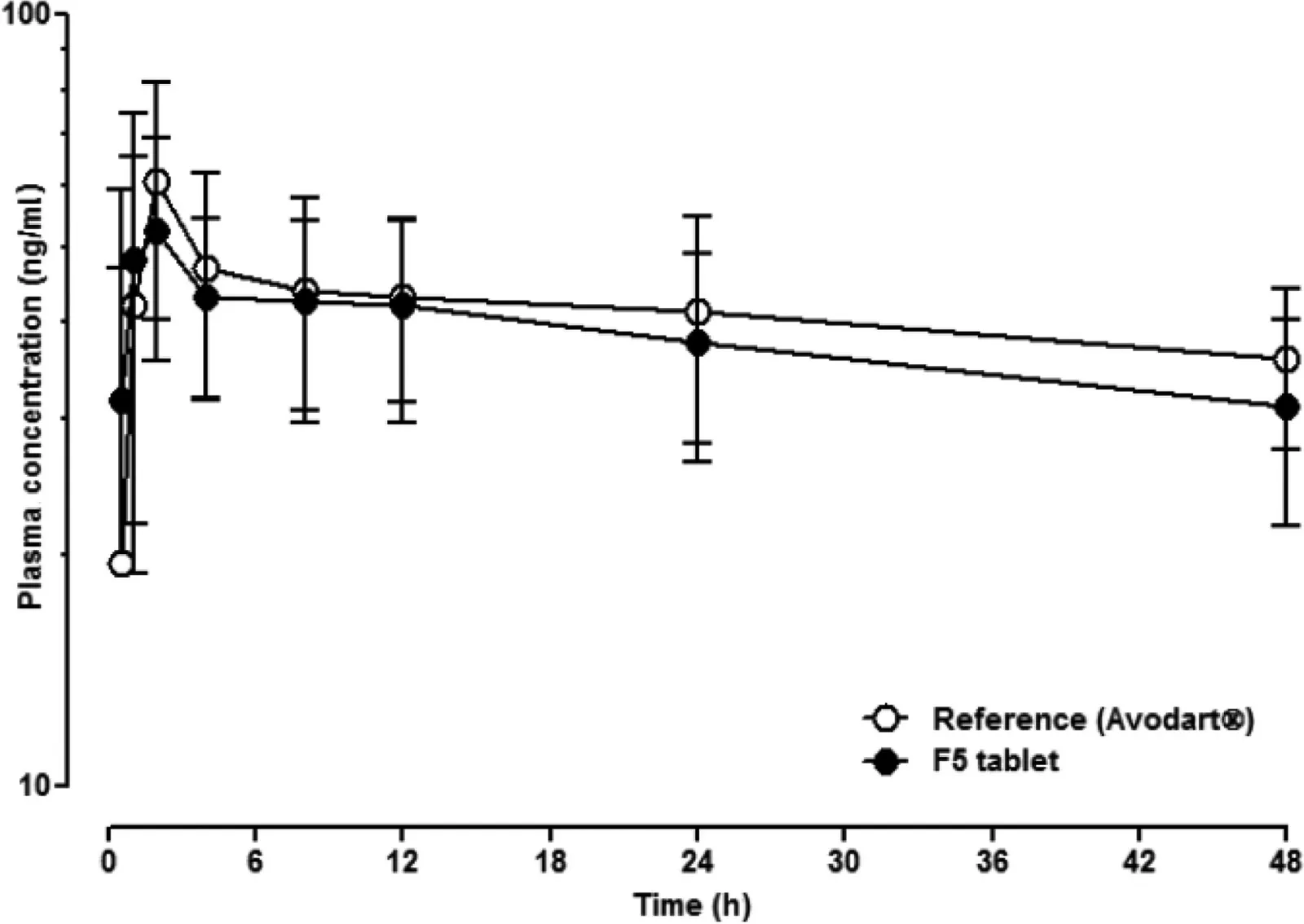
Fig.5-Mean plasma concentration-time profiles of dutasteride after oral administration of the reference soft gelatin capsule(Avodart®)or the F5 tablet in beagle dogs(n=6,crossover).Each point and vertical bar represent the mean and standard deviation,respectively.
4. Conclusion
Duγ CD-PS complexes were successfully prepared by the simple oven-drying method.The amorphous form of dutasteride was confirmed via DSC and pXRD studies. In vitro dissolution of dutasteride from the tablets of Duγ CD-PS complexes was comparable with that from the reference (Avodart®,soft gelatin capsule). Moreover, in vivo pharmacokinetic parameters of the Duγ CD-PS complex tablet after oral administration in beagle dogs were not significantly different from that of the reference. These results suggest the feasibility of developing a tablet formulation of dutasteride with bioequivalence to the commercial soft gelatin capsule,which requires further evaluation in a larger number of animals and/or humans.
Conflicts of interest
The authors report no conflicts of interest.The authors alone are responsible for the content and writing of this article.
Acknowledgments
We appreciate Dr. Nam-Doo Kim at the New Drug Development Center (Daegu-Kyeongbuk Medical Innovation Foundation, South Korea) for conducting a computer simulation study of the dutasteride-cyclodextrin complex.This research was supported by the National Research Foundation of Korea(NRF),funded by the Korean government(MSIP)(No.2009-0083533).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.ajps.2018.08.007.
猜你喜欢
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Recent strategies on targeted delivery of thrombolytics
- Cellulose based polymers in development of amorphous solid dispersions
- Phyto-phospholipid complexes(phytosomes):A novel strategy to improve the bioavailability of active constituents
- MSNCs and MgO-MSNCs as drug delivery systems to control the adsorption kinetics and release rate of indometacin
- Effects of granulation process variables on the physical properties of dosage forms by combination of experimental design and principal component analysis
- Percutaneous absorption and brain distribution facilitation of borneol on tetramethylpyrazine in a microemulsion-based transdermal therapeutic system