微乳毛细管电动色谱-场放大富集法测定9种核苷类化合物
2019-08-30于晓章梁美娜李宁杰聂谨芳黄丽丽
张 庆,于晓章,张 琳,梁美娜,李宁杰,聂谨芳,黄丽丽
(1.桂林理工大学环境科学与工程学院,广西桂林541006;2.广西环境污染控制理论与技术实验室,广西桂林541006;3.福州大学食品安全与药物化学教育部重点实验室,福建福州361006;4.桂林理工大学化学与生物工程学院,广西桂林541006)
在基因工程、免疫学等现代生物技术的推动下,核苷及其衍生物等得到了广泛的使用。尤其是核苷类化合物,其在抗病毒、抗肿瘤方面发挥了越来越重要的作用。目前,经美国食品药品监督管理局(FDA)批准的抗艾滋病药物大多是核苷类衍生物,因此核苷类药物有望成为新一代具有重要作用的药物。目前,核苷类化合物的分析检测方法主要有高效液相色谱法、薄层色谱法、柱色谱法及毛细管电色谱法等[1-5]。高效液相色谱法不仅分析时间长,而且流动相消耗大量的有机溶剂;薄层色谱法仅适用于少量样品的分析;柱色谱法常需要加压操作,不利于大规模样品的测定;毛细管电色谱法具有快速、高效的特点,但存在重现性较差,运行时易产生气泡等难题。因此,亟需探索和发展一种更为高效的分析介质和更为灵敏的检测手段,才能更进一步发掘核苷类药物在生物医学中的作用。
微乳液毛细管电动色谱(MEEKC)是以微乳液作为分离介质,根据物质的疏水性以及电迁移率的差异来实现分离的一种毛细管微分析技术,该技术可以同时对电荷型/中性物质、水溶性/脂溶性物质进行检测[6-8]。MEEKC法具有高效、快速及分析对象广等优点,但由于其检测器多为紫外检测器(UV),受到进样量和光程等因素的影响,灵敏度低,因此限制了MEEKC法在痕量分析中的应用。为了提高该分析方法的检测灵敏度和扩大其应用范围,一些在线富集技术,比如场放大富集(FASI)、大体积样品堆积(LVSS)和离子选择性耗尽扫集法等已被用于 MEEKC[9-10]。Zhang等[11]建立了一种 MEEKC-FASI分析尿液中吗啡、可待因、纳洛酮、海洛因、蒂巴因、可卡因、哌替啶、芬太尼、美沙酮9种麻醉剂的方法,该技术检出限低至0.3μg/L。Chen等[12]首次建立了一种基于毛细管内衍生化的胶束电动色谱法(MEKC)测定奶粉、液态奶、奶饮料和豆奶粉样品中羟脯氨酸的方法。在最佳条件下,该方法可在7min内完成衍生分离,脯氨酸的检出限为1.6ng/mL。
本研究建立了一种新的MEEKC-FASI分析方法,对影响MEEKC分离效果和FASI富集效果等因素进行了优化,实现了腺嘌呤、鸟嘌呤、甲基腺苷、N6-甲基腺苷、胞苷、鸟苷、次黄嘌呤、巯嘌呤、氟尿嘧啶9种核苷类化合物的同时在线分析检测。为了验证所建立的方法的可行性,进行了尿样和血清样品的加标回收率实验,回收率范围85.2%~113.0%,方法检出限低至0.22μg/mL。
1 实验部分
1.1 主要仪器及试剂
安捷伦 HP3D毛细管电泳仪(Agilent technologies Inc),配备二极管阵列检测器;KQ-100型超声清洗仪(昆山超声仪器有限公司);pHS-3C型精密酸度计(上海大普仪器有限公司);R200D分析天平(Sartorius);800型离心沉淀器(上海手术器械厂)。未涂层熔融石英毛细管(63cm,有效长度54.5cm,50μm i.d.×375μm o.d.)。
腺嘌呤(Aderine)、鸟嘌呤(Guanine)、甲基腺苷(Methladenosine)、N6-甲基腺苷(N6-methyladenosine)、胞苷(Cytidine)、鸟苷(Vernine)、次黄嘌呤(Hypoxanthine)、巯嘌呤(Mercaptopurine)、氟尿嘧啶(Fluorouracil)标准品(中国药品生物制品鉴定所);十二烷基硫酸钠(SDS)(Alfa Aesar);色谱纯甲醇、乙腈(国药集团化学试剂有限公司);实验用水均为Milli-Q超纯水。
血清和尿液(南京森贝伽生物科技有限公司)。
1.2 标准溶液和微乳液的配制
标准溶液:分别称取适量的腺嘌呤、鸟嘌呤、甲基腺苷、N6-甲基腺苷、胞苷、鸟苷、次黄嘌呤、巯嘌呤、氟尿嘧啶标准品,用超纯水溶解,配制成1 000μg/mL的标准储备溶液,避光保存于4℃冰箱。使用时再稀释至所需的浓度。微乳液:分别取13.5mg的SDS,11.0μL的正丁醇,8.0μL的乙酸乙酯和1470.0μL 10mmol/L的Na2B4O7缓冲溶液,置于2mL离心管中,超声30min即可得到光学透明稳定的微乳缓冲体系。固定微乳组成为质量分数为0.9%SDS、0.6%的正丁醇、0.5%的乙酸乙酯和98.0%10mmol/L的Na2B4O7缓冲溶液。
1.3 样品制备
尿样:取适量浓度的9种核苷类化合物标准溶液,混匀,加入尿液中,经4 000r/min离心10min后取上层清液,用0.22μm滤膜过滤,再用10mmol/L Na2B4O7溶液稀释10倍,制得尿样。储存在4℃冰箱备用。血清样品:取适量浓度的9种核苷类化合物标准溶液混匀,加入血清中,经离心处理10min(4 000r/min)后,取上层清液用0.22μm滤膜过滤,再用10mmol/L Na2B4O7溶液稀释20倍,制得血清样品。储存在4℃冰箱备用。
1.4 样品分析
毛细管预处理:依次用超纯水、0.1mol/L HCl、超纯水、0.1mol/L NaOH、超纯水各冲洗30min,最后在电泳仪上用运行缓冲液冲洗30min。待毛细管电泳(CE)基线和电流稳定后,方可进行CE操作分析。CE仪器操作:毛细管每次使用前依次用超纯水、0.1mol/L NaOH溶液、超纯水和缓冲液各冲洗10min;两次运行之间,依次用超纯水和运行缓冲液冲洗5min;每运行5次,均需及时更换运行缓冲液;每次更换运行缓冲液后,均需用缓冲液平衡毛细管20min。所有溶液进入CE仪器运行前,均需经0.22μm滤膜过滤,并超声2min脱气。安捷伦HP3D毛细管电泳仪检测波长设置为200nm,柱温保持为25℃。
2 结果与讨论
2.1 MEEKC分离条件的优化
2.1.1 缓冲溶液pH的选择 缓冲溶液的pH值对 MEEKC的分离效果有显著的影响[10-12]。考察了Na2B4O7缓冲溶液的pH在8.0~9.5之间变化时对9种核苷类化合物的分离情况。结果表明随着pH值增大,分析时间逐渐延长。如图1所示,当pH为8.0和8.5时,9种核苷类化合物不能实现基线分离;当pH为9.5时,N6-甲基腺苷(4号峰)和胞苷(5号峰)不能基线分离。综合考虑分离度和迁移时间,选择缓冲溶液的最佳pH值为9.0。
2.1.2 缓冲溶液浓度的选择 使用低浓度(5~10mmol/L)的硼砂盐或磷酸盐能够获得较小的电流和较大的电渗流,因此上述2种缓冲溶液常被应用于MEEKC体系中[13-14]。固定缓冲溶液的pH值为9.0,考察浓度5~15mmol/L Na2B4O7缓冲溶液对9种核苷类化合物分离效果的影响。如图2所示,随着硼酸盐浓度的增加,分离度逐渐增大,被分析物的迁移时间也随之延长,并且在15mmol/L的时候系统产生的焦耳热变大,导致体系的稳定变差。综合考虑,缓冲溶液浓度选择10mmol/L。

图1 Na2B4O7缓冲溶液pH值对9种核苷类化合物分离效果的影响Fig.1 Effect of Na2B4O7buffer pH on the separation of 9nucleosidesseparation voltage:15kV;temperature:25 ℃;detection wavelength:200nm;electrokinetic injection:10kV×6s;microemulsion:0.9%SDS,0.6%1-butanol,0.5%ethyl acetate and 98.0%10mmol/L Na2B4O7buffer solution.1.aderine(2μg/mL);2.guanine(2 μg/mL);3.methladenosine (2.5 μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL).

图2 NaB4O7缓冲溶液浓度对9种核苷类化合物分离效果的影响Fig.2 Effect of NaB4O7buffer concentration on the separation of 9nucleosidesborate concentration:5,10and 15mmol/L;Other conditions were the same as Fig.1.1.aderine(2μg/mL);2.guanine(2μg/mL);3.methladenosine(2.5μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL).
2.1.3 表面活性剂的选择 SDS是MEEKC法中最常用的阴离子表面活性剂。实验研究了浓度为5~20mmol/L SDS对核苷类化合物分离效果的影响[11,15]。结果显示,随着SDS浓度的增大,由于电渗流的减小以及油滴表面负电荷的增多,迁移时间相应的延长。当SDS的浓度为15mmol/L时,次黄嘌呤(7号峰)和巯嘌呤(8号峰)迁移时间比较接近,分离度较差;当SDS的浓度为20mmol/L时,虽然基线分离较好,但迁移时间较长;当SDS的浓度较低时(5mmol/L),9种分析物分离度较差,腺嘌呤(1号峰)和鸟嘌呤(2号峰)甚至未基线分离。考虑微乳体系需要足够的表面活性剂来维持稳定,最终选择10mmol/L的SDS为最佳浓度。
2.1.4 助表面活性剂的选择 正丁醇是MEEKC法中最常用的助表面活性剂[11,16-17]。微乳体系中加入一定浓度的正丁醇,可以增加微乳滴液膜的机械强度,有利于维持MEEKC运行时微乳液的稳定。但如果正丁醇过量,多余的正丁醇分子会与表面活性剂(如SDS)的极性基团缔合,导致微乳液膜松散,降低微乳体系的稳定性,容易出现破乳现象[9]。因此,考察了正丁醇含量对9种核苷类化合物分析效果和微乳液稳定性的影响。结果显示,正丁醇质量分数由0.3%増至0.9%时,随着正丁醇含量的增加,迁移时间随之延长;但正丁醇质量分数增到1.2%,迁移时间缩短,且分离度很差。综合考虑各分析物的分离度和灵敏度,选择0.6%的正丁醇为最佳助表面活性剂。
2.1.5 油相种类及浓度的选择 油相一般采用正己烷、正庚烷、正辛烷、乙酸乙酯等化合物[11,18]。鉴于乙酸乙酯与水之间的表面张力更小,因此将其作为微乳体系的油相微乳液,仅需少量的表面活性剂即可维持体系的稳定。以乙酸乙酯为油相,考察了其浓度对9种核苷类化合物分离效果的影响。在0.25%~0.75%(质量分数)乙酸乙酯含量范围内,9种分析物均完全分离,且选择性和灵敏度均无明显的变化。为确保微乳体系能在较低的SDS浓度下维持稳定,故确定0.5%乙酸乙酯为油相。
2.1.6 分离电压的选择 分离电压不仅影响分离效率,也是影响分析时间的一个重要参数[11,18-24]。研究了在10、15、20、25kV的条件下,分离电压对9种核苷类化合物分离效果和灵敏度的影响。结果发现,随着分离电压的增大,9种分析物的迁移时间逐渐缩短。当电压为10kV时,分析物的分离时间较长,且巯嘌呤(8号峰)和氟尿嘧啶(9号峰)响应值都有所下降;当电压为20kV和25kV时,分析物的分离度均有所下降,原因可能为体系的电流较大导致毛细管焦耳热较大,引起电流和基线不稳定。综合考虑,实验选择15kV为最合适的分离电压。综上所述,确定最佳的实验条件为:微乳液组成为质量分数0.9%的SDS、0.6%的正丁醇、0.5%的乙酸乙酯和98.0%的10mmol/L Na2B4O7缓冲液(pH=9.0),分离电压为15kV。9种核苷类化合物在最佳微乳组成条件下的MEEKC谱图如图3所示。从图中可以看出,除了N6-甲基腺苷(4号峰)和胞苷(5号峰)分离度相对较小外,9种分析物在12min内基本实现基线分离。
2.2 富集条件的优化
2.2.1 样品稀释剂的选择 FASI主要基于高压电场下样品溶液和背景缓冲溶液电导的差异而实现样品富集的,因此样品基体对FASI富集效果有重要的影响[7,23-24]。分别以10mmol/L Na2B4O7缓冲溶液、微乳液、0.1mmol/L NaOH溶液、甲醇和水为样品稀释剂,考察了样品稀释剂对9种核苷类化合物灵敏度和分离度的影响。结果发现,当样品稀释剂为微乳液时,虽然鸟嘌呤的响应值明显得到提高,但其余分析物的分离度下降,氟尿嘧啶甚至不出峰;0.1mmol/L NaOH溶液作为样品稀释剂时,腺嘌呤响应值下降,N6-甲基腺苷峰变宽;甲醇为样品稀释剂时,鸟嘌呤、巯嘌呤和氟尿嘧啶响应值下降;水为样品稀释剂对各分析物的灵敏度显著下降。在10mmol/L Na2B4O7缓冲溶液中,9种核苷类化合物的灵敏度均得到提高,且具有良好的分离度。综合考虑,选择10mmol/L Na2B4O7缓冲溶液作为样品稀释剂。
2.2.2 进样条件的选择 场放大进样技术的进样量主要取决于进样电压和进样时间。考察进样电压在14~24kV范围内9种核苷类化合物的灵敏度变化情况。随着进样电压的增大,检测灵敏度也随之提高;当进样电压超过22kV时,各分析物的峰形展宽,分离度开始下降,说明此时样品富集失败。原因可能为:(1)FASI富集过程中,进样量随进样电压(或者进样时间)的增大而增大,当进样量超过毛细管所能承受的范围时(FASI进样量只能低于毛细管总体积的10%),由于样品扩散作用,导致峰形展宽,灵敏度和分离度下降;(2)高进样电压使样品区带产生大量的焦耳热,引起毛细管内温度骤升,影响体系电流和基线的稳定;(3)样品稀释剂和背景缓冲溶液存在电导差异,过高的进样电压,可能引起体系产生气泡,导致富集失败。进样电压为22kV,考察进样时间对峰高的影响。进样时间从5s增加到15s的过程中,被分析物的峰高仅在5~10s的范围内是随着进样时间的增加而增高。当进样时间为15s时,峰形展宽,分离效率下降,故以10s(22kV)为最佳进样时间。
2.2.3 FASI与常规进样对比及富集倍数 文献报道[11,25]在样品电动进样前压力注入一段水塞可以提高富集效果。实验结果表明,当以3kPa压力进6s水柱时,富集效果最好。在最佳的MEEKC条件下,对比了FASI和常规进样两种方式对9种核苷类化合物分离选择性和检测灵敏度的影响。如图4所示,在不影响核苷类化合物分离度和迁移时间的情况下,采用FASI进样方式可显著提高各分析物的检测灵敏度。
对比常规进样方式,场放大富集的倍数可表示为:f=(h/h0)×(c0/c)。其中,h0和h分别为常规进样和采用FASI进样后溶质的峰高,c0和c分别为两种情况下溶质的浓度。依据公式可以计算出9种核苷类化合物的富集倍数分别为:腺嘌呤(33倍)、鸟嘌呤(12倍)、甲基腺苷(38倍)、N6-甲基腺苷(14倍)、胞苷(18倍)、鸟苷(20倍)、次黄嘌呤(21倍)、巯嘌呤(8.8倍)及氟尿密度(14倍)。

图3 最佳MEEKC条件下9种核苷类化合物的电泳谱图Fig.3 Electropherogram of 9nucleosides under the optimized MEEKC conditionsseparation voltage:15kV;temperature:25 ℃;detection wavelength:200nm;electrokinetic injection:10kV×6s;microemulsion solution:0.9%SDS,0.6%1-butanol,0.5%ethyl acetate and 98.0%10mmol/L Na2B4O7buffer solution.1.aderine(2μg/mL);2.guanine(2μg/mL);3.methladenosine(2.5μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL).
2.3 线性范围和检出限
配制一系列不同浓度的9种核苷类化合物的混合标准溶液,在最佳富集条件下进行MEEKC测定。各分析物的线性范围、线性回归方程、相关系数及检出限(S/N=3)如表1所示。9种核苷类化合物在线性范围内相关系数大于0.9960,相对标准偏差(RSD)均小于5.74%。
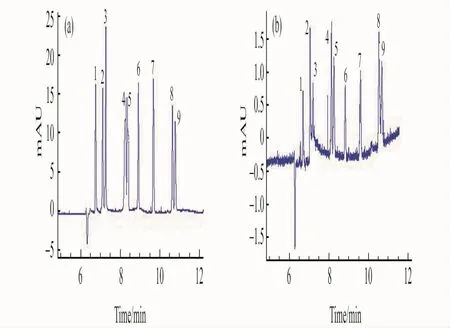
图4 MEEKC-FASI(a)与常规MEEKC(b)进样的9种核苷类化合物毛细管电泳谱图Fig.4 Elecropherograms of 9nucleosides for MEEKC-FASI(a)and conventional MEEKC (b)(a):MEEKC-FASI:water plug(30mbar,6s);electrokinnetic injection(22kV×10s).1.aderine(2μg/mL);2.guanine(2μg/mL);3.methladenosine(2.5μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL);(b):normal electrokinetic injection,10kv×5s;1.aderine(2μg/mL);2.guanine(2μg/mL);3.methladenosine(2.5μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL).
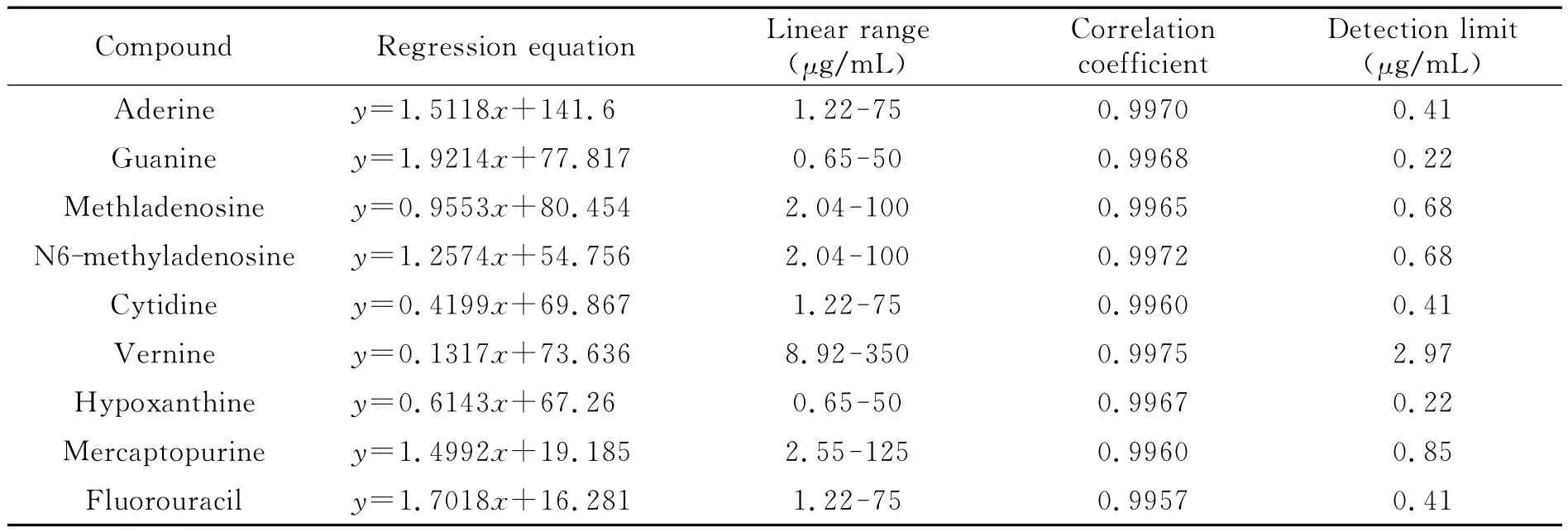
表1 9种核苷类化合物的线性回归方程、线性范围、相关系数和检出限Table 1 Regression equations,linear ranges,correlation coefficients and detection limits for 9nucleosides
2.4 回收率和精密度
2.4.1 尿样的测定 根据1.3节方法制备尿样,按照1.4节所述的条件进行分析,尿样的电泳谱图如图5所示。由图5可以看出,除了N6-甲基腺苷(4号峰)和胞苷(5号峰)峰形有略微展宽,分离度变小,以及巯嘌呤(8号峰)和氟尿密度(9号峰)的分离度有所下降以外,其他分析物均没有受到基质的影响。分别向尿样中加入2个不同浓度水平的9种核苷类化合物的标准混合溶液进行加标回收率实验。每个浓度平行测定3次,计算相对标准偏差(RSD)。如表2所示,9种核苷类化合物的回收率介于91.2%~113.0%之间,RSD均小于5.9%。加标回收实验结果表明,所建立的方法稳定可靠。

图5 9种核苷类化合物尿样(a)和空白(b)的电泳谱图Fig.5 Elecropherograms of 9nucleosides in urine(a)and in blank samples(b)experimental conditions same as the Fig.4(a).1.aderine(2μg/mL);2.guanine(2μg/mL);3.methladenosine(2.5μg/mL);4.N6-methyladenosine(5μg/mL);5.cytidine(4μg/mL);6.vernine(30μg/mL);7.hypoxanthine(5μg/mL);8.mercaptopurine(4μg/mL);9.fluorouracil(2μg/mL).
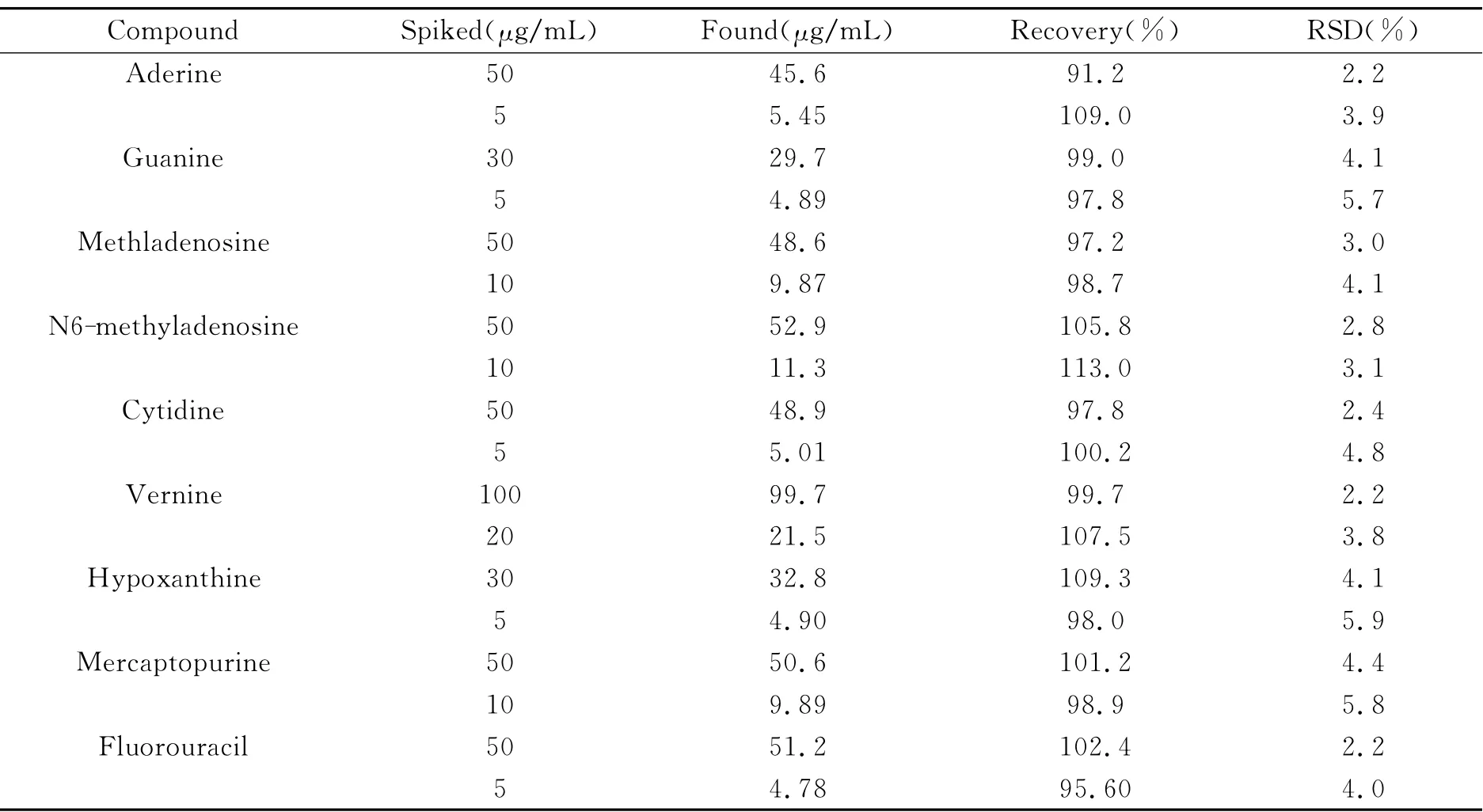
表2 尿样中9种核苷类化合物的加标回收率(n=3)Table 2 Recoveries of 9nucleosides spiked in urine samples(n=3)
2.4.2 血清样品的测定 血清样品根据1.3节步骤处理,按照1.4节所述的条件进行分析。同时为验证所建立的方法应用于血清样品中9种核苷类化合物检测结果的可靠性,进行了2个浓度水平的加标回收率实验。每个浓度平行测定3次,计算相对标准偏差(RSD),得到9种核苷类化合物的回收率介于85.2%~111.8%之间,RSD均小于8.2%。
3 结论
本文建立了一种快速、灵敏的同时测定9种核苷类化合物的微乳液毛细管电动色谱-场放大富集分离分析方法,分别研究了MEEKC分离条件、FASI富集条件等因素的影响。在最佳的实验条件下,9种核苷类化合物可在12min内实现基线分离,且被测物浓度与峰电流呈良好的线性关系,检出限(S/N=3)低至0.22μg/mL。此外,将所建立的方法应用到尿样和血清样品分析,成功地检测到了9种核苷类化合物。该方法具有简单、灵敏等特点,有望在药物分析等领域得到广泛的应用。
