NLRP3炎性小体在牙周疾病中的研究进展
2019-08-14朱博文陈立妹郭竹玲
朱博文 陈立妹 郭竹玲,2
1.海南医学院口腔医学院 海口 570100;
2.海南医学院第一附属医院口腔科 海口 570100
牙周病是发生于牙周支持组织的慢性炎性疾病。全球范围大数据统计得出,20%的成年人可由牙周炎导致的骨缺损不可逆地走上失牙之路[1]。牙周病的病程进展被认为是由内源性革兰氏阴性菌感染引发和维持的炎症性宿主反应的结果。
炎性小体(inflammasome)是一组蛋白质复合物,是宿主免疫反应的关键参与者。其在机体中能够识别包括病原相关分子模式(pathogen-associated molecular pattern,PAMP)和损伤相关分子模式(damage-associated molecular pattern,DAMP)的多种诱导刺激,是细胞内重要的先天免疫传感器;当先天性免疫防御受损时,活化的受体同样可以参与适应性免疫防御[2-4]。NLRP3炎性小体是核苷酸结合寡聚化结构域样受体家族含热蛋白结构域亚成员,是炎性小体众多家族成员中研究最广泛、深入的一个,已被证实在牙周病的发生、发展中扮演关键角色。
1 NLRP3炎性小体
自2002年Martinon首次提出炎性小体的概念,世界各地的研究者便对炎性小体展开各方面的研究,并将其与宿主防御以及各类疾病联系起来。
Bostanci等[5]证实,NLRP3炎性小体表达水平显著增高与牙周疾病关系密切。到目前为止,已经在人体22种不同的核苷酸结合寡聚域(nucleotide-binding oligomerization domain,NOD)样受体(NOD-like receptor)/核苷酸结合域与含富亮氨酸重复序列受体(nucleotide-binding domain and leucine-rich repeat-containing receptor,NLR)家族蛋白质分子以及热蛋白结构域(Pyrin domain,PYD)的NLRP中鉴定出14种亚家族成员(NLRP1~NLRP14)[6-8]。
NLRP3炎性小体是由3种组分组成的,即NLRP3、细胞凋亡相关斑点样蛋白(apoptosisassociated speck-like protein containing a caspaserecruitment domain,ASC)以及半胱氨酸天冬氨酸特异性蛋白酶(cysteinyl aspartate specific proteinases,caspase)-1组成。
NLRP3包括3个特征性结构域,即C端富亮氨酸重复序列(leucine-rich repeat,LRR)、中央NACHT(NAIP、CIITA、HET-E和TP1)以及N端PYD。ASC由其N端的PYD和C端的caspase活化募集结构域(caspase-activating and recruitment domain,CARD)组成[3-4,9]。NLRP3可通过PYD结合含PYD-CARD的接头蛋白ASC,并以CARD结构域募集并触发caspase-1激活,从而促进白细胞介素前体(pro-interleukin,pro-IL)-1β和pro-IL-18成熟,分泌白细胞介素(interleukin,IL)-1β与IL-18[4](图1)。IL-1β与IL-18是牙周病发生、发展过程中重要的促进因子。
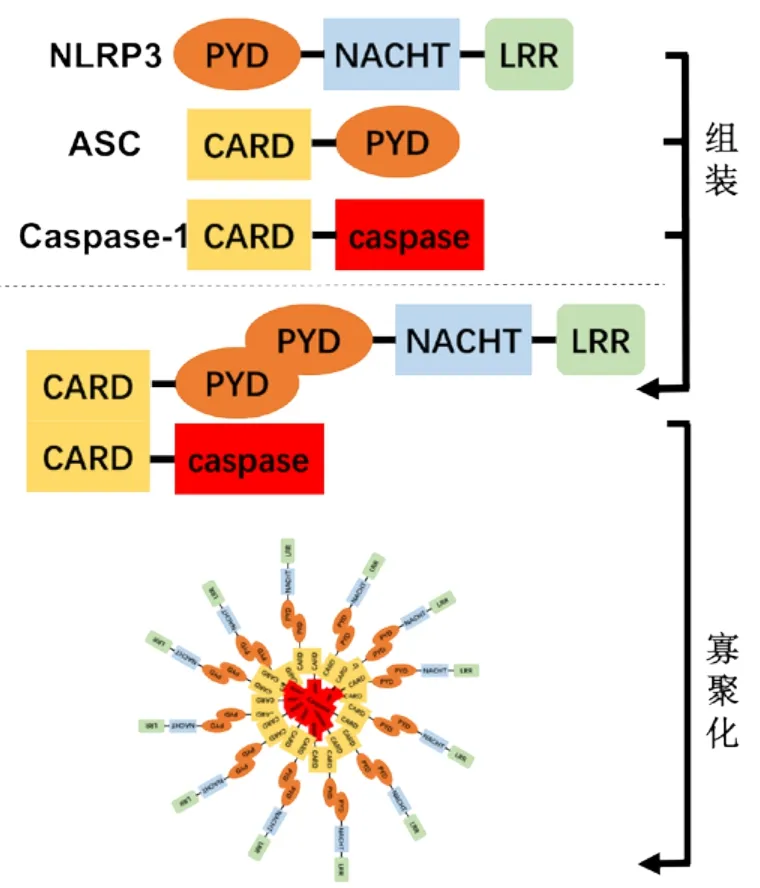
图 1 NLRP3炎性小体的组成和组装Fig 1 Composition and assembly of NLRP3 inf l ammasome
2 NLRP3炎性小体在牙周疾病中的信号传导机制
NLRP3炎性小体与炎症性牙周疾病联系紧密[5],NLRP3炎性小体可以引发牙龈上皮细胞早期宿主免疫应答。慢性牙周炎与广泛型侵袭性牙周炎患者牙周组织中NLRP3的表达显著高于健康组织[10],Isaza-Guzmán等[11]的研究也显示,唾液中NLRP3、ASC和IL-1β水平与慢性牙周炎和广泛型侵袭性牙周炎关系密切。
通常有2个信号参与NLRP3炎性小体活化(图2):1)信号一为微生物刺激或内源性细胞因子,如脂多糖(lipopolysaccharide,LPS)等被Toll样受体(Toll-like receptor,TLR)或NLR识别,并以肿瘤坏死因子(tumor necrosis factor,TNF)-α、核因子(nuclear factor,NF)-κB等炎性因子依赖性方式诱导NLRP3表达的上调,同时促进pro-IL-1β、pro-IL-18的产生[4,12];2)信号二来自不同种类的NLRP3炎性小体激活剂,数量繁杂,主要包括外源性微生物刺激、内源性危险信号以及无机晶体结构3类[13]。一旦激活,这些复合物将促进caspase-1活化,以及NLRP3炎性小体寡聚化,并将pro-IL-1β和pro-IL-18裁剪成具有活性的IL-1β和IL-18[4,9,12]。
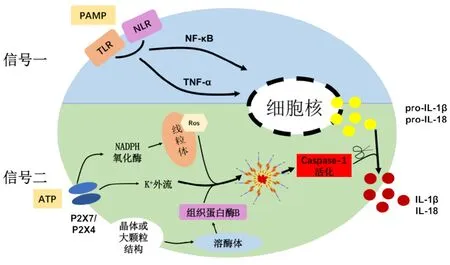
图 2 NLRP3炎性小体的信号传导模型Fig 2 Signaling models of NLRP3 inf l ammasome
在人巨噬细胞中,牙龈卟啉单胞菌(Porphyromonas gingivalis)可以通过阻断Fn介导caspase-1激活及IL-1β的分泌。Taxman等[14]认为第二信号是研究NLRP3炎性小体受牙周微生物抑制的新思路。目前有3种模型得到普遍认同:离子流动模型、活性氧(reactive oxygen species,ROS)模型以及溶酶体破裂模型[4,7,13](图2)。这3种模型并不是相互独立与排斥,而是互相呼应与补充。Shimada等[15]提出3种模型将汇集同一途径来诱导释放氧化的线粒体DNA,以激活NLRP3炎性小体的统一理论。
2.1 离子流动模型
在离子流动模型中,K+等离子水平的变化在NLRP3炎性小体激活中起关键作用,如细胞外三磷酸腺苷(extracellular adenosine triphosphate,eATP)激活离子通道P2X7并触发快速K+流出,对NLRP3具有活化作用[4]。正如Almeida-da-Silva等[16]所回顾,已证实牙周疾病中P2X7和P2X4受体对于炎性小体NLRP3的激活作用,牙龈上皮细胞表面P2X7受体可作为组装NLRP3炎性小体的第二个信号,不仅对eATP诱导的IL-1β分泌至关重要,而且对细胞内pro-IL-1β处理也很重要。研究[17-19]证实,K+流出是牙龈卟啉单胞菌介导的NLRP3炎性小体激活所必需的。P2X7受体所介导的第二信号传导同样也是NLRP3炎性小体受抑制的作用点。Morandini等[20]的研究发现,牙龈卟啉单胞菌的菌毛能够通过干扰嘌呤受体P2X7的ATP信号传导,来减少P2X7活化所介导的pro-IL-1β加工,在第二信号水平抑制IL-1β。
2.2 ROS模型
ROS形式的氧化应激广泛地涉及NLRP3炎性小体活化,所有炎性小体NLRP3刺激物都将引发ROS的产生[4,7]。在牙龈上皮细胞中,发现ATP通过由P2X4/P2X7/pannexin-1组成的复合物诱导ROS产生,而生成的ROS能够激活NLRP3炎性小体和caspase-1[19,21];在P2X7受体传导ATP刺激的过程中,还原型烟酰胺腺嘌呤二核苷酸磷酸(reduced form of nicotinamide-adenine dinucleotide phosphate,NADPH)氧化酶和线粒体会协同生成线粒体ROS(mitochondrial ROS,mROS)[22],而抑制NADPH氧化酶也降低了ATP诱导的mROS产生[19]。如果ROS生成的途径被阻断,NLRP3炎性小体也会受到抑制。Choi等[22]发现,牙龈卟啉单胞菌效应物核苷二磷酸激酶(nucleosidediphosphate kinase,NDK)可以阻断牙龈上皮细胞中eATP刺激P2X7受体并由NADPH氧化酶活化介导产生ROS氧化应激传导链的过程,并抑制ATP介导的炎性小体活化过程中caspase-1激活及IL-1β的分泌[23]。使用NDK同源物也可抑制eATP刺激引发的先天性免疫反应[23]。
2.3 溶酶体破裂模型
溶酶体破裂模型是针对晶体或大颗粒结构激活剂(如尿酸晶体、明矾、二氧化硅、疟疾疟原虫、羟磷灰石等)时所提出的。吞噬细胞吞噬这些激活剂导致溶酶体损伤,NLRP3炎性小体能够感知外漏的溶酶体内容物[4,7]。Montenegro Raudales等[24]发现未烧制的牙结石激活的NLRP3炎性小体依赖于吞噬作用,并且牙结石的晶体结构刺激NLRP3炎性小体的活化并且部分地促进人和小鼠吞噬细胞中的IL-1β分泌。该模型进一步提出了组织蛋白酶B是NLRP3炎性小体所感知的介质[4,7]。Park等[18]发现组织蛋白酶B的抑制显著降低牙龈卟啉单胞菌诱导的细胞毒性和ASC胞吞体形成的程度,也会降低了牙龈卟啉单胞菌诱导的caspase-1活化和IL-1β生成。牙龈卟啉单胞菌能在一定程度上影响细胞的运输,并通过逃避通向溶酶体的内吞途径来抑制NLRP3炎性小体。Taxman等[14]认为这种抑制效果是牙龈卟啉单胞菌调节核心细胞骨架蛋白的结果,而通过抑制内吞作用可鉴定抑制炎性小体的牙龈卟啉单胞菌成分,这有利于牙周炎病因的探索与治疗的开发。
3 NLRP3基因多态性在牙周疾病中的免疫调控
在牙周疾病中,不仅NLRP3炎性小体可以引发牙龈上皮细胞早期宿主免疫应答,巨噬细胞同样也可被NLRP3炎性小体所激活[25]。牙龈卟啉单胞菌可抑制巨噬细胞内单钠尿酸盐、肽聚糖和明矾诱导的NLRP3炎性小体活化,但却不抑制ATP和尼日利亚菌素诱导的NLRP3炎性小体活化[14]。而这种NLRP3炎性小体的抑制与活化现象在牙周疾病中并不单一。以慢性牙周炎中最常见的病原体牙龈卟啉单胞菌为例,其LPS刺激牙龈上皮细胞上调IL-1β表达和促进细胞内IL-1β积累[26],与eATP刺激一并被认为是NLRP3炎性小体活化的关键[27]。但在某些状态下,牙龈卟啉单胞菌也可抑制NLRP3炎性小体活化。在牙龈卟啉单胞菌感染的人髓样单核细胞系Mono-Mac-6细胞中,牙龈卟啉单胞菌通过下调NLRP2和ASC表达来减少NALP3炎性复合体的表达[5],而在牙龈上皮细胞中,牙龈卟啉单胞菌感染可直接下调牙龈上皮细胞中NLRP3的表达,但对ASC的表达无显著影响[26]。报道[28]表明即使NLRP3的表达升高,牙龈卟啉单胞菌可直接诱导NLRP3的蛋白质水解。
在复杂的口腔生态环境、多种细菌共生的条件下,多数学者支持龈上生物膜可增加牙龈纤维细胞中caspase-1、ASC、AIM2、IL-1β和IL-18的表达,但不影响NLRP3表达。龈下生物膜在较低浓度下增强caspase-1、ASC、AIM2、IL-1β和IL-18表达,在较高浓度下将其下调,均可对NLRP3表达产生显著影响[29-30]。牙周病原体影响NLRP3炎性小体活化与抑制这2种截然不同状态,被认为是其操控宿主先天免疫反应的途径。通过活化NLRP3炎性小体分泌IL-1β与IL-18,可显著增加牙槽骨损失,破坏牙周组织形成适合牙周病原体繁殖的环境[31];当宿主对细菌免疫反应过强时,可通过抑制NLRP3炎性小体降低宿主的反应强度,便于细菌繁衍与入侵[14]。
另有研究[29,32]表明,牙龈卟啉单胞菌具有协同感染功能,可策略性干扰多种炎症组分的表达,实现操纵先天性免疫反应的目的,进而维系牙周组织中相关微生物物种的存活和持久性。
牙周病原体影响NLRP3炎性小体的活化与抑制,可能与NLRP3基因多态性与基因突变有关。目前的免疫遗传学研究[33-34]表明,牙周病发病机制具有个体差异,而宿主遗传因素是影响个体易感性重要的因素。NLRP3是细胞内“危险信号”的传感器,NLRP3基因多态性可致NLRP3炎性小体过度活化。不同个体面对外源性感染会引发成慢性炎症反应[35]。
临床中也常见即使有大量牙菌斑、牙石形成但仍然保持牙周健康的患者;也有牙周生物膜积累较少但极易患牙周疾病的个体[33-34]。Isaza-Guzmán等[36]的研究表明,NLRP3(rs4612666)CT/CC基因型的单核苷酸多态性与衰老和/或吸烟的协同相互作用在牙周病的致病途径中起重要作用;还发现NLRP3炎性小体突变可导致人类遗传性炎性疾病的发生。cryopyrin相关周期热综合征是一种常见的自身免疫性疾病,以皮疹、关节痛和长时间的发热为特征,NLRP3突变导致caspase-1活化增强和自发性IL-1β和IL-18分泌增加,而IL-1抑制剂可有效地控制病情进展。NLRP3在常染色体隐性遗传性疾病掌跖角化-牙周破坏综合征发生、发展中也发挥着重要作用[37],并与克罗恩病、银屑病幼年特发性关节炎、白塞综合征和类风湿性关节炎在内的若干炎症性疾病易感性显著相关[36]。NLRP3(第705位谷胱酰胺)和CARD8(第10位半胱苷酸)的变异被认为可能是常见慢性炎症性疾病的重要因素[35,38],NLRP3的功能获得性突变可导致NLRP3组成性活化,导致IL-1R依赖性的IL-1β过表达[35]。IL-1β单核苷酸多态性也与牙周病存在相关性。
4 展望
目前,对于NLRP3在牙周疾病中活化与抑制的现象已得到确认,就其发生机制已提出部分假说。确切的机制以及各假说之间的关系尚未明确。多种复杂牙周微生物对NLRP3炎性小体的综合调节作用还存在诸多空白。对于NLRP3炎性小体基因多态性与基因突变在牙周疾病中的研究也刚刚起步。探索NLRP3炎性小体的多重功能,有助于为临床治疗牙周病与相关慢性炎性疾病提供新思路;了解NLRP3炎性小体基因与牙周疾病易感性的关系有利于牙周疾病易感人群的确定与牙周疾病的预防。明确抑制NLRP3炎性小体的机制有利于对慢性炎性疾病治疗方法的研发。
