极长链酰基辅酶A脱氢酶缺乏症1例并文献复习
2019-07-30孙小兰车圆圆章静静钟建民
孙小兰,车圆圆,章静静,钟建民
(江西省儿童医院神经内科,南昌330006)
极长链酰基辅酶A脱氢酶缺乏症(very long chain acyl-CoA dehydrogenase deficiency,VLCADD)是一种罕见的常染色体隐性遗传病[1],是可导致脂肪酸β氧化过程受阻、供能障碍及中间代谢产物蓄积的一组疾病。该病最早由BERTRAND等[2]于1993年首次报道,其发病率低,江西省儿童医院2016年收治1例VLCADD患儿,现将其临床资料及相关文献回顾性分析如下。
1 临床资料
患儿,女,2岁10个月。因呕吐、嗜睡1 d于2016年1月18日入本院。患儿于起病前1 d晚上醒后突发呕吐胃内容物4~5次,未予特殊处理,次日患儿呈嗜睡状态,呼叫不醒,仍有呕吐,无发热、抽搐、腹泻和发绀,于当地医院查血糖0.9 mmol·L-1,给予高渗糖静脉推注后转本院。入院后查体:T 37.7 ℃,P 114 次·min-1,R 26 次·min-1,体质量14 kg。嗜睡状态,呼吸平稳,双侧瞳孔等大等圆,对光反射灵敏。口唇红润,颈稍抵抗。心肺腹查体阴性。四肢肌张力、肌力正常,生理反射存在、病理反射未引出。入院后复查血糖正常,电解质正常,C肽、胰岛素均正常,丙氨酸转氨酶正常,天冬氨酸转氨酶升高,肌酸激酶升高,肌酸激酶同功酶正常,乳酸升高,血氨正常。脑脊液常规及生化正常。脑电图提示清醒睁眼全脑各区1~2 Hz,50~60 μV δ活动。头颅磁共振提示两侧脑室体后部旁白质内异常信号,提示髓鞘化延迟。心电图正常。入院诊断为中枢神经系统感染,予阿昔洛韦抗感染、补液等治疗,患儿神志转清,精神反应好转,无呕吐。血串联质谱检测显示肉豆蔻酰肉碱(C14)、肉豆蔻烯酰肉碱(C14:1)、肉豆蔻二烯酰肉碱(C14:2)和棕榈烯酰肉碱(C16:1)均升高,尿有机酸气相质谱检测显示戊二酸、已二酸、辛二酸、庚二酸等多种二羧酸尿症(表1—2),符合VLCADD代谢改变。5 d后好转出院,出院时患儿神志清楚,无呕吐,无抽搐,精神正常,出院诊断为VLCADD。
2017年10月4日因呕吐、精神差1 d,昏迷数小时再次入住本院重症监护室(PICU)。病程中2次出现心脏骤停,予心肺复苏后好转,复苏后出现抽搐1次,表现为神志不清,牙关紧闭,口唇发绀,四肢强直阵挛,予推注地西泮后缓解。入院前当地血糖0.6 mmol·L-1,入院后查血糖正常,电解质正常,丙氨酸转氨酶正常,天冬氨酸转氨酶升高,肌酸激酶升高,肌酸激酶同功酶升高,乳酸升高,血氨正常。复查血尿代谢病筛查,结果与第一次检测相仿(表1—2)。入院后予监测血糖,保护脏器、静脉营养支持治疗,治疗8 d后好转出院。嘱家属注意患儿饮食,增加喂食次数,缩短喂养间隔时间(白天间隔约4 h,夜间间隔约10 h)[3],随访至今患儿除活动量减少、睡眠增多外,余与正常儿童无异。
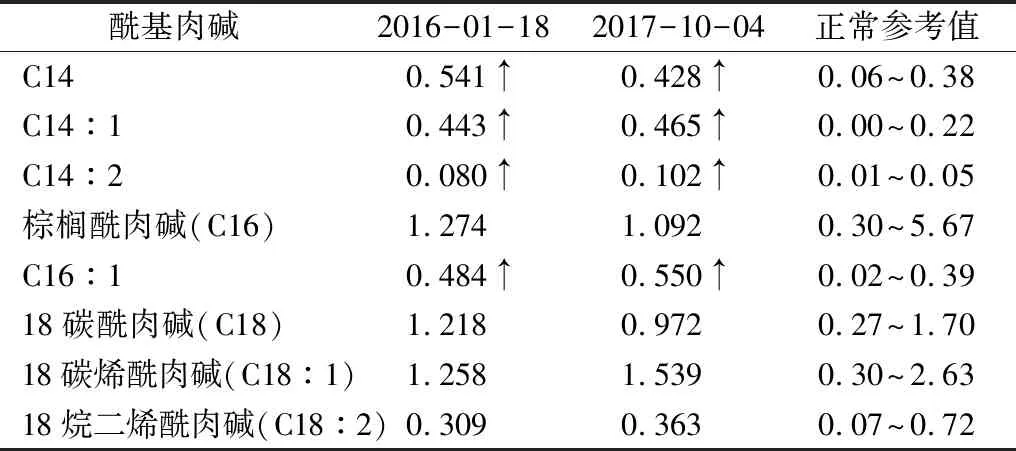
表1 2次入院血串联质谱检测结果 c/(μmol·L-1)

表2 2次入院尿有机酸气相质谱检测结果 c/(μmol·L-1)
2 讨论
VLCADD的发病率尚不明确,欧美国家新生儿筛查示检出率约为1/85 000[4]。亚洲发病率相对较低[5],国内曹金俊等[5]对8年间在上海新华医院儿科就诊的11例VLCADD的临床表现、实验室检查、基因型、诊治和预后等进行分析,其他以散在报道[6-13]多见。
极长链酰基辅酶A脱氢酶(VLCAD)存在于人体心肌、骨骼肌、肝脏、成纤维细胞等组织,是线粒体脂肪酸β氧化中第一步的关键酶,催化含14~18碳的不同长度碳链的脂酰基辅A脱氢其辅酶为黄素腺嘌呤二核苷酸,由黄素腺嘌呤二核苷酸接受脱氢产生的氢原子进入线粒体呼吸链进行氧化磷酸化产生ATP,同时脂肪酸β氧化过程中还可产生乙酰辅酶A,参与三羧酸循环,或通过肝脏形成酮体,在运动、饥饿、应激等情况下产生能量,为骨骼肌、心肌和肝脏等重要器官提供能量来源[14]。VLCAD缺陷将导致体内长链脂肪酸代谢障碍,长链脂肪酸不能氧化供能,并且在骨骼肌、心肌、肝脏、成纤维细胞中蓄积,对细胞产生毒性作用。
2.1 VLCADD分型
VLCADD主要分为3型。1)心肌病型:最为常见,主要在新生儿和婴儿早期发病,起病早且凶险,病情重,病死率极高,主要表现为低酮症性低血糖、瑞氏综合征、新生儿猝死、肥厚型和扩张型心肌病、心包积液、心率失常、肌酸激酶水平升高。2)肝病型:临床相对少见,多于婴儿晚期或幼儿期起病,该型症状较轻,常表现为反复发作的低酮性低血糖,可伴有肝功能异常,很少伴有心肌损害,但若诊断和治疗不及时,也有可能危及生命。3)肌病型:为迟发型,主要在青少年至成年期发病,表现为运动、感染、饥饿后横纹肌溶解和肌红蛋白尿,甚至可发生肾功能衰竭,可伴有肌无力、痛性肌痉挛或肌痛等[2,14]。本例患儿2岁10个月起病,反复发作性低血糖,伴有肝功能轻度异常,符合肝病型表现,2次起病时均表现为呕吐后出现嗜睡,第一次发作后血糖0.9 mol·L-1,肝功能轻度异常,予高糖及对症治疗后,迅速好转;第二次起病家属没有足够重视,未及时送医,致使病情进展迅速,出现心脏骤停和抽搐情况。提示,肝病型病情比心肌病型轻,若诊治不及时,同样可能危及生命。
2.2 VLCADD的诊断
诊断VLCADD有赖于串联质谱检查,C14:1常作为VLCAD缺陷症的识别标记。LIEBIG等[15]认为,血串联质谱进行新生儿疾病筛查时,若C14:1大于1 μmol·L-1即可诊断为VLCADD;对于C14:1高于正常,但低于1 μmol·L-1的患儿,则应高度怀疑该病可能。本例患儿2次检测C14:1高于正常,分别为0.443、0.465 μmol·L-1,提示该病可能性大。本例患儿己二酸、辛二酸、癸二酸等多种二羧酸明显升高,结合临床表型,VLCADD的诊断成立。提示,如果尿有机酸气相质谱分析发现二羧酸尿症,可进一步支持本病,但值得注意的是,轻症患者或伴有横纹肌溶解的患者可无二羧酸尿症。
目前,基因检测水平已非常成熟,VLCADD是常染色体隐性遗传病,致病基因为ACADVL(OMIM 609575)[10],位于染色体17p13.1,长约5.4 kb,内含20个外显子,编码655个氨基酸的多肽。目前已发现166种基因突变类型,错义突变为主要的突变类型,但无热点基因,无义突变可导致VLCAD完全失去活性,而错义突变或单个氨基酸的缺失可使VLCAD留有残余部分酶活性,症状较轻[16]。一般基因检测仅有80%病例可以籍此明确诊断,VLCADD患儿的ACADVL基因突变谱具有高度异质性,新生儿型和婴儿型较成人迟发型患者病死率高,提示基因型和表型间存在一定相关性[5]。孟卫京等[8]认为,遗传代谢病筛查实验结合高通量基因测序诊断对VLCADD的早发现、早干预,提升疗效和患儿生活质量有重要意义。本例患儿基因结果未发现已知致病突变,这可能是一种尚未被认知的新的突变类型或内含子突变或调控基因缺陷等。
2.3 VLCADD的治疗
VLCADD的治疗原则是给予高碳水化合物和低脂饮食,限制长链脂肪酸的摄入,辅以中链甘油三酯(medium-chain triglycerides,MCT),避免饥饿,维持血糖稳定,补充肉碱,对长链脂肪酸代谢产物进行监测,加强护理,避免感染、劳累及饥饿,并进行相应的对症支持处理。增加喂食次数可作为一种简单有效的预防措施,可达到避免饥饿、维持血糖的目的。肉碱的补充治疗,目前仍存在争议,短期应用可以促进酮体生成和减少空腹低血糖的发生,但过多则会促进长链酰基肉碱的生成和蓄积,对机体产生毒性作用,故动态监测体内肉碱及酰基肉碱水平有助于调整治疗方案中肉碱的供给及脂肪配比,对于监测治疗效果,改善预后有极其重要的作用[3]。本例患儿遵医嘱增加喂养次数,在合并感染或过度运动时及时补充葡萄糖后预后良好,这可能与肝病型VLCADD症状相对较轻有关。
总之,VLCADD是先天性脂肪酸代谢缺陷疾病,早诊断、早治疗是关键[3,5,13]。对不明原因的反复呕吐、肝功能、心肌酶异常,特别是出现低酮性低血糖时要尽早进行遗传代谢病筛查,尤其在疾病发生的急性期,若高度怀疑不能明确诊断时可反复多次送检。肉豆蔻烯酰基肉碱(C14:1)升高,可作为诊断VLCADD最重要的代谢指标,二羧酸尿症,可进一步支持肝病型的诊断,如有条件应行基因或酶学检测以进一步明确诊断。另外,应尽可能避免脑损伤,提高患儿的生存率和生存质量,及时确诊先证者有助于提供遗传咨询和产前诊断,从而达到优生优育的目的。
