食源性致病菌等温扩增检测技术的研究进展
2019-07-27吴朦晨张明洲俞晓平
葛 航,吴朦晨,张明洲,俞晓平
(中国计量大学 生命科学学院 浙江省生物计量及检验检疫技术重点实验室,浙江 杭州 310018)
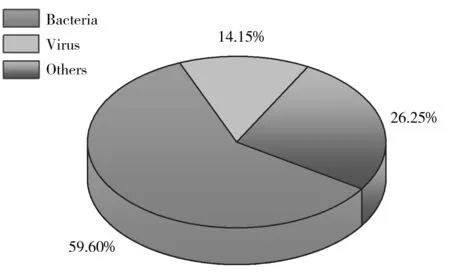
图1 世界范围内食源性风险因子危害程度分析[1]Fig.1 Global burden of diseases caused by food-borne factors[1]the percentage was calculated by dividing number of deaths caused by certain factor over total number of deaths
微生物与食品安全有着密切的关系,有益微生物被广泛应用于食品制造与加工过程;有害微生物则会导致食品腐败变质或直接危害人类健康,引起食品安全事件。世界卫生组织最新的食源性疾病调查报告显示,全世界因食源性风险因子致病的人数超过6亿人,死亡人数超过41万人,其中因细菌导致死亡的人数超过24万人,占总死亡人数的59.60%[1](图1)。据国家卫生计生委统计,2011~2016年我国因微生物引起的食物中毒事件为2 446起,占食物中毒事件的25.5%,致病人数达4.82万人,占总致病人数的43.3%,其中以肠炎沙门氏菌(Salmonellaenteritidis)和副溶血弧菌(Vibrioparahaemolyticus)为代表的食源性致病菌是引发食品安全事件的主要因子[2]。2018年8月25日发生在桂林的百人以上集体食物中毒事件,就是因参会人员食用了被沙门氏菌污染的食物所致。对食源性致病菌进行现场或在线快速精准检测是保障公众安全的重要手段,无论是对突发性食品安全事件的快速响应,或是对高危害性致病菌的有效防控,都需要相应的快速检测方法作为支撑。然而,细菌微小的个体和复杂的种类,使得对其准确鉴定具有一定的困难,这也凸显了开发快速、准确而又高效的致病菌检测方法的必要性和迫切性。
1 食源性致病菌核酸检测方法
微生物的传统鉴别方法(培养法)一般基于其形态和生化特性,需要经过选择性增菌、分离菌株、扩增培养、制片观察形态等常规生化鉴定和毒素鉴定步骤。培养法虽然判定准确性高、稳定性强,但耗时通常为5~6天,且对检验者的经验和技巧要求较高。因此,培养法在一些需要做出快速反应或检测量大的现场或在线检测中局限较大。除此之外,有些细菌能够进入不可培养状态(Viable but non culturable,VBNC),即菌体具有活性但无法被培养,这种情况会导致所检测的细菌数量被低估[3]。因此,基于形态观察和生化鉴定的传统方法虽然可作为最终的仲裁方法,却无法在有限的时间内完成对大量样品的筛查,恰恰相反,这正是以聚合酶链式反应(Polymerase chain reaction,PCR)技术为代表的致病菌核酸检测方法所具备的优点。
PCR方法以细菌特异性核酸序列作为检测目标,简化了判断细菌种类的标准,并通过指数级的扩增,提高了检测灵敏度。PCR技术的结果非常可靠,一方面,细菌的特征基因具有非常高的特异性,在其他细菌种类中可能不存在或者序列不同,即使是种间较为保守的基因,如16S基因,也在不同种类间具有多样性的特定区域,序列完全相同的可能性极低;另一方面,通过DNA测序技术,能够获得详细的序列碱基组成和排列信息,为判断是否检测到靶标基因提供可靠证据。同时,以核酸为靶标的检测方法检测通量高、检测时间短,在检测效率上有着明显的优势。由于PCR的以上诸多优点,相关研究在近十年呈现快速增长的趋势(图2),在国标中一些致病菌的检测方法也采用了PCR方法(表1)。
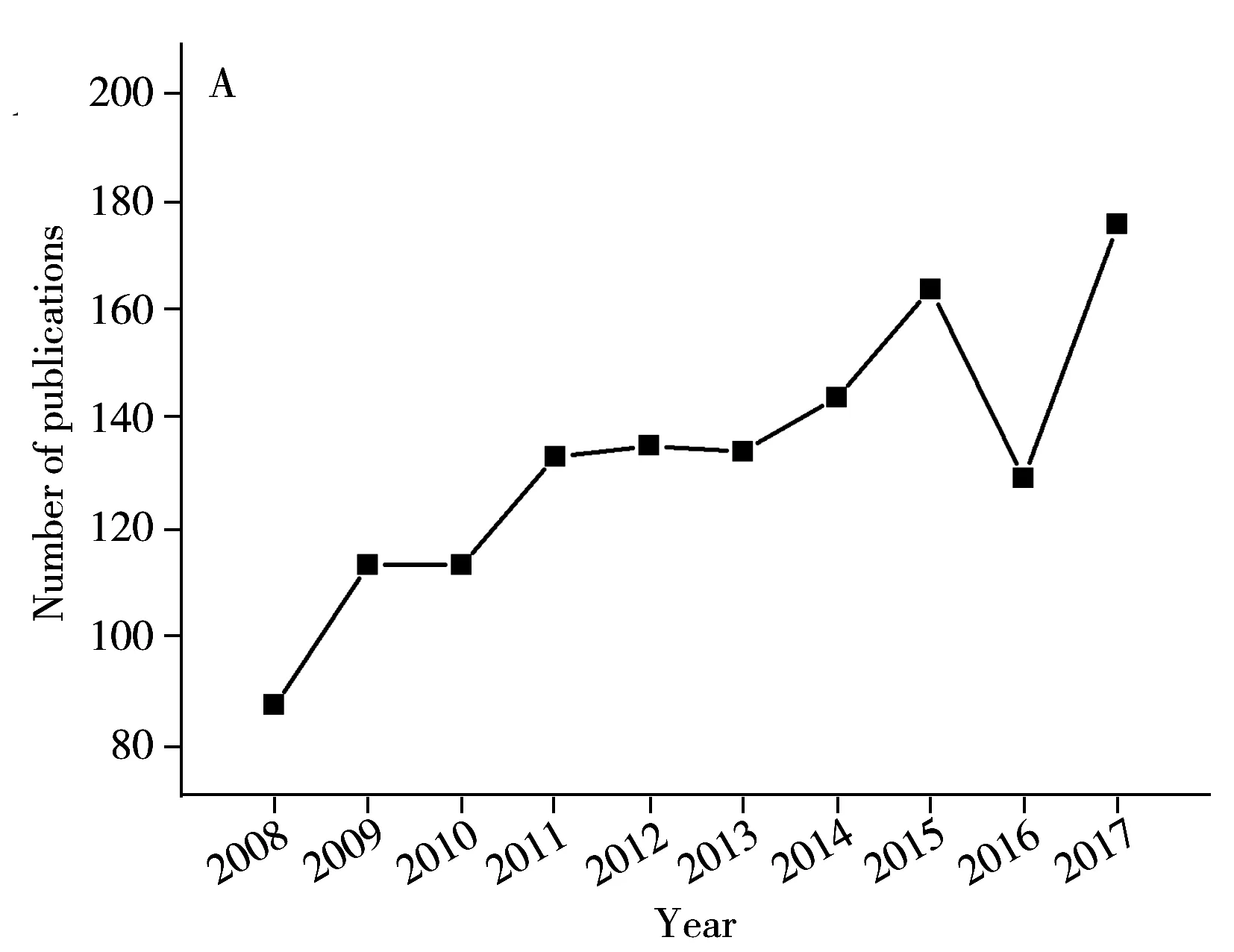
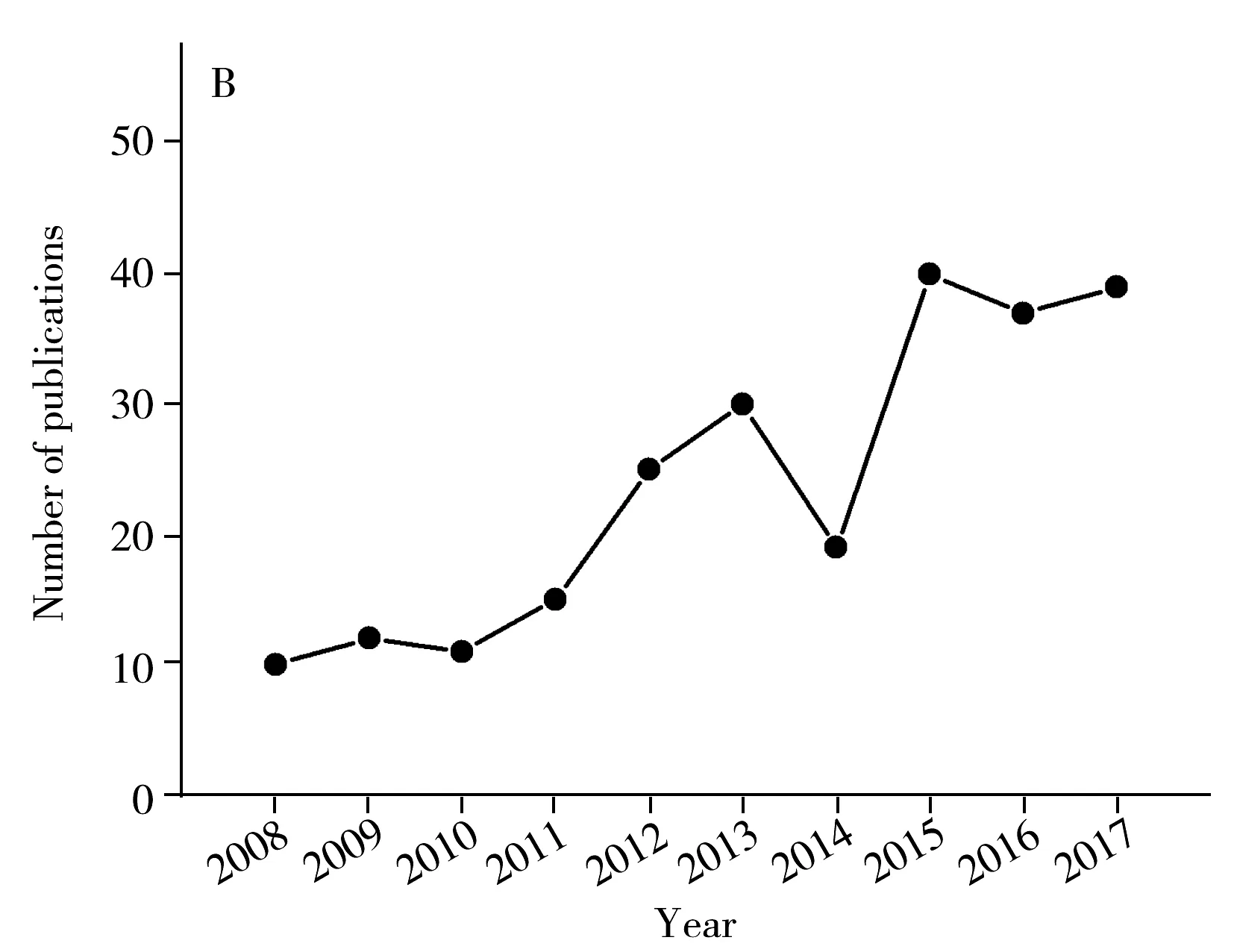
图2 沙门氏菌PCR检测方法研究情况Fig.2 Publications of Salmonella spp.detection methods targeting specific genes(data were derived from Web of Science)A.PCR based methods for Salmonella spp.(沙门氏菌核酸扩增方法研究数量);B.isothermal amplification based methods for Salmonella spp.(沙门氏菌等温扩增方法研究数量)
2 等温扩增技术
经典的PCR方法能够在几小时内完成对目标核酸的指数级扩增,具有检测时间短、适用范围广的优势,但也存在明显的缺陷。PCR过程需要经历变性、退火、延伸3个步骤的多次循环,而且每一个步骤都需要在特定的温度下进行,通常分别为94、55、72 ℃。变温循环要求PCR仪器配备快速变温单元,导致仪器不便移动和携带,而实际应用中又存在着大量现场或在线检测的需求,由此催生了多种可在恒温条件下进行的PCR技术,称为等温扩增技术,如环介导等温扩增(Loop-mediated isothermal amplification,LAMP)、滚环扩增(Rolling circle amplification,RCA)、链置换扩增(Strand displacement amplification,SDA)、切口酶信号扩增(Nicking enzyme signal amplification,NESA)和核酸外切酶Ⅲ辅助扩增(Exonuclease Ⅲ assisted amplification)技术等[4-6],详见表2。这些技术不需要复杂的温度变化,在一个恒定温度下就可以实现对检测目标或者信号的扩增,极大地降低了对仪器的需求,具有现场应用或在线检测的潜力。

表2 常见5种等温扩增方法比较Table 2 Comparison of 5 isothermal amplification methods
2.1 环介导等温扩增技术(LAMP)
LAMP技术是目前最为常用的病原菌现场检测技术,在2000年首次提出[7]。与传统PCR不同,LAMP技术针对模板6个不同区域的位置,设计可以自发形成环状结构的引物,在不断复制中将线性的目标DNA序列无限拓展为之字形的长链结构。尽管LAMP的反应过程非常复杂,但是结果的判断却简便、可视。这是由于反应产生的大量焦磷酸离子与镁离子形成了难溶于水的焦磷酸镁沉淀,使得肉眼即可定性判别检测结果。但更精确的定量判断则需要借助于浊度计或使用能识别DNA双链的核酸染料[8]。目前,研究人员已经成功地将LAMP技术应用于多种致病微生物的检测中,包括单增李斯特菌(Listeriamonocytogenes)[9-11]、沙门氏菌(Salmonellaspp.)[12-14]、空肠弯曲杆菌(Campylobacterjejuni)[15-17]、产志贺毒素大肠杆菌(EscherichiacoliO157)[18]、副溶血性弧菌(Vibrioparahaemolyticus)[19-21]和霍乱弧菌(Vibriocholerae)[22-24]等。LAMP方法扩增速度快、灵敏度高、无需变温,在所有等温扩增方法中应用最广。但是,LAMP的扩增产物组成复杂,多种长度片段共存的产物起不到再次验证扩增特异性的作用;此外,60 ℃的反应温度仍然需要装置维持;极快的扩增速率也增加了假阳性的风险。这些缺点是LAMP技术所亟待解决的。

图3 滚环扩增技术(RCA)原理示意图Fig.3 Principle of rolling circle amplification
2.2 滚环扩增技术(RCA)
RCA的原理与滚环复制有很多相似之处,滚环复制是噬菌体和病毒常见的环状双链DNA复制方式,由具有3端至5端外切酶活性的DNA聚合酶Ⅲ在切口处同步进行新链合成和原有链去除[25]。RCA技术只使用单侧引物,结合在环形待检测的核酸序列上,在聚合酶的作用下,不断循环复制,形成不断重复并且与环形模板互补的DNA长链[26];待测核酸非环状类型时,可将锁状引物(Padlock probe)与待测线型核酸结合,使用DNA连接酶连成环形,以锁状引物的序列代替核酸序列进行扩增,进而使用相应策略检测引物的数量(图3)。Zhou等[27]巧妙地将一小段双链结构加入到锁状引物中,形成哑铃型。由于双链结构的存在,RCA扩增的长链DNA中也会形成多个双链结构,在反应体系中加入嵌入型荧光染料,即可对双链结构的数目做出估计,达到对目标核酸序列定量分析的目的。Guo等[28]将RCA与氯高铁血红素的氧化还原反应结合,用于大肠杆菌(Escherichiacoli)的定量测定。Teng等[19]先用抗体与适配体混合的夹板式方法捕获目标菌体,再通过RCA技术放大信号,实现了对鱼肉中副溶血弧菌的精准测定。由于锁状引物的序列是人为设计的,因此RCA的产物序列具有很强的拓展性,可以根据实际需要来调整,以便形成具有信号产生功能或者催化活性的DNA高级结构,也易于实现指数级扩增。此外,RCA的产物在空间上是连续的,因此是唯一可进行原位扩增的等温扩增策略,特别适合在芯片表面进行非均相体系扩增。但是,线性单链DNA需与锁状引物进行连接反应后再开始扩增反应,这决定了RCA容易受到复杂溶液体系的干扰,所以RCA在均相体系中的检测效果不如非均相体系[28]。

图4 链置换扩增技术(SDA)原理示意图Fig.4 Principle of strand displacement amplification
2.3 链置换扩增技术(SDA)
SDA是一种通过建立循环置换机制实现扩增特定核酸序列的方法,与LAMP相比,SDA对反应温度要求低,在室温条件下也能进行(图4)。SDA通常产生较短的DNA单链,与嵌入型荧光染料、Tagman探针或分子信标(Molecular beacon,MB)等结合,在扩增目标序列的同时放大检测信号,为定量检测提供基础。SDA单链的产生可以由具有3端至5端外切酶活性的DNA聚合酶引发[29],也可以由基于热力学稳定性的Toehold置换引发[30-31]。由于产物为单链DNA,具备识别其他核酸序列的能力,因此SDA也是实现DNA逻辑运算的常用手段[32]。通常情况下,SDA中的靶标在启动一次置换反应后,无法被回收,因而只能实现线性扩增,但在切口酶(Nicking enzyme)帮助下,也可实现指数级的快速扩增[33]。Tan等[34]将两段与目标序列互补的DNA通过切口酶的结合序列串联起来,以碱基配对原则与目标序列结合,在聚合酶作用下形成平末端DNA双链后,切口酶结合到新形成的双链识别原件上,此时可剪开一个缺口,释放刚合成的单链(目标序列),用于下一轮循环;与此同时,被切口酶切割的双链结构可在聚合酶作用下重新形成目标序列,达到指数级扩增效果。然而,指数级SDA需要多个DNA结构单元配合,体系较为复杂,容易受到干扰;SDA反应的触发需要DNA单链,无法直接对双链DNA扩增。这些缺点限制了SDA的大范围应用,目前尚无基于SDA的商业化检测产品。
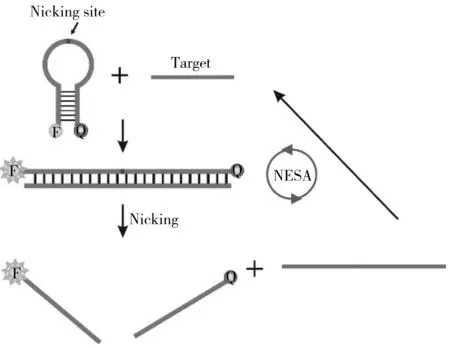
图5 切口酶扩增技术(NESA)原理示意图Fig.5 Principle of nicking enzyme signal amplification

图6 核酸外切酶扩增技术原理示意图Fig.6 Principle of exonuclease Ⅲ assisted amplification
2.4 切口酶信号扩增技术(NESA)
切口酶(Nicking enzyme)与限制性内切酶相似,能够识别特异的核酸序列,但切口酶只催化一条链的磷酸二酯键断裂。切口酶的这一特性主要用于均相检测中核酸靶标的回收以及信号的产生。切口酶介导的扩增体系由靶标识别元件、信号产生元件、切口酶识别序列组成,通过切口酶切割信号探针并回收靶标用于下一轮信号产生(图5)。Li等[35]设计了一种特殊的分子信标,信标的环形部分碱基序列与需要检测的目标序列互补,并且其中含有切口酶的识别序列。信标与目标序列互补后产生的双链结构,在热力学稳定性上要高于分子信标的茎环结构。因此,当目标序列出现在检测体系中时,会使分子信标的构象发生改变,打开原来的茎环结构,形成新的双链DNA结构,进而被切口酶切割。切割后,双链之间的氢键数目显著减少,稳定性大大降低,在55 ℃的条件下会自动解离,释放目标序列,用于结合下一个分子信标,形成扩增循环。目前已经有采用类似策略检测肠炎沙门氏菌(Salmonellaenteritidis)16S rRNA序列的报道[36]。切口酶等温扩增方法的最大困难在于酶切识别位点的引入。通常情况下,核酸靶标中不含有识别序列,需要结合其他策略来生成,这限制了NESA的应用[37]。NESA更适合作为SDA的辅助单元,配合进行信号的二次放大[38]。
2.5 核酸外切酶Ⅲ辅助扩增技术
除了切口酶,核酸外切酶Ⅲ(Exonuclease Ⅲ)也能通过类似的策略,达到持续放大信号的目的[39]。核酸外切酶Ⅲ具有3端到5端脱氧核糖核酸外切酶活性,与切口酶相比,核酸外切酶不需要特定的识别序列即可工作,并且只作用于DNA双链的平末端或者3端方向凹陷的粘性末端,因此适用面更广(图6)。Zuo等[40]对常规的分子信标结构做了些许调整,在3端猝灭基团后增加了若干碱基,形成凸出型的3端粘性末端,以避免信标本身被核酸外切酶Ⅲ降解。当信标结合目标DNA序列形成双链结构时,3端变为平末端类型,碱基逐个被核酸外切酶剥离,荧光基团也因为猝灭基团的远离而发光。当剩余的碱基数量不足以维持稳定的双链结构时,5端含有荧光基团的短链解离,释放目标序列进行下一个循环。Luo等[41]将捕获探针固定在电极表面,利用核酸外切酶Ⅲ持续地降解与目标结合的捕获探针,使得电极表面的DNA数量持续减少,导电性增加,据此来检测常见的肠道细菌特征DNA序列。核酸外切酶Ⅲ只识别粘性末端类型而不区分DNA序列的特点,便于在非均相体系中同时降解不同序列的DNA双链,所以主要用于固相材料捕获靶标DNA后信号的产生。特别当与石墨烯等纳米材料结合时,核酸外切酶Ⅲ可以快速释放荧光基团,从而解除石墨烯对荧光的猝灭效应,产生信号[42]。但是,经核酸外切酶Ⅲ降解后的单链DNA分解为单个游离核苷酸,无法进行回收,因此难以实现循环,一般只能实现信号的线性扩增。
3 等温扩增存在的共性问题
等温扩增技术在近十多年间的发展非常迅速,LAMP、RCA、NESA、SDA等核酸高效扩增方法的出现,有效地缩短了检测所需时间,也降低了对复杂检测设备的依赖,为致病微生物的现场实时检测提供了良好的基础。目前,等温扩增技术已经成为致病微生物快速检测的重要方法之一,常见的食源性致病菌,均有众多成熟的等温扩增检测方法报道,以及基于等温扩增原理开发的商业化试剂盒[43-46]。但随着人们对食品安全问题关注程度的逐渐提高,以及政府部门监管力度的逐渐加强,致病菌检测的需求急剧增长,并且出现了不少新变化和新问题。对此,等温扩增方法还需要在一些共性问题上加以改进,以适应这些实际需求。
3.1 活性识别
等温扩增所检测的物质是致病菌DNA,其性质相对稳定,细菌死亡后DNA不会立即降解,因此仍可以被扩增手段检测到。严格意义上来说,等温扩增所得到的结果,只能证明样品中细菌DNA的存在,而无法获知细胞的生存状态,这可能导致对细菌危害风险的误判。
3.2 非特异性扩增
快速是等温扩增的关键特点,也是实现其高灵敏度的重要原因。然而,短时间内对目标基因的高速率复制,在显著提升方法灵敏度、缩短检测时间的同时,也大大增加了等温扩增对于非靶标DNA的敏感性,导致非特异性扩增概率上升,产生假阳性。例如,在LAMP操作过程中,只要反应体系与外界空气有过短暂接触,就非常容易受到气溶胶污染,导致扩增产物中含有大量的非目标基因片段。
3.3 前处理步骤
复杂的溶液成分会影响氢键的稳定性,从而降低引物对靶基因的识别效率。因此,样品的前处理对于等温扩增技术来说非常关键。细菌DNA必须经过富集、提取和纯化,减弱其他因素的干扰,才能被高效识别和扩增,否则将会引起背景值偏高或信号值减弱,导致信噪比降低。由于前处理主要依赖人工操作,在某些情况下,前处理所消耗的时间已经超过了等温扩增所需时间,成为等温扩增进一步快速化的制约因素[47]。
3.4 检测通量
为了进一步提高等温扩增的效率,更好地适应现场或在线检测,不少研究都致力于在单个等温扩增反应中实现多个核酸靶标的检测,即多重等温扩增策略[48](图7)。但是,单纯通过增加多组引物达到同时扩增多个目的片段的方式具有较大局限性。增加引物组数必然导致体系的复杂化,不同组引物之间存在交叉组合的可能,增加了获得非特异性条带的几率。目前,多重等温扩增策略能同时检测的靶标一般为2到3种,3种以上的多重反应往往由于引物组之间的干扰导致灵敏度不佳。
3.5 解决措施
在细菌活性区分问题上,研究发现,提取DNA前用叠氮溴化丙锭(Propidium monoazide,PMA)处理细菌,可以实现对活菌的选择性扩增。死亡细菌通常会丧失对细胞壁通透性的调控能力,使得PMA可以自由通过,并结合死亡细菌核区内的基因组DNA,阻止其被DNA聚合酶结合[13]。基于此原理,马骉等[49]构建了PMA-LAMP方法,成功区分了样品中死亡的和具有活性的副溶血弧菌。但是,在某些情况下,细胞壁的通透性并不能实时反映细菌的活性,此时PMA并不能很好地发挥区分活性的作用[50]。
在非特异性扩增问题上,张明洲等[51]优化了钙黄绿素、氯化锰配比及反应液镁离子浓度,降低了染料对于酶扩增效率的影响,从而省略了扩增完成后加入染料的步骤,使得污染几率显著降低。也有研究者从LAMP产物的识别角度出发,利用分子信标的特异性识别功能,只对目标序列产生信号[52],虽然在一定程度上降低了假阳性率,但是污染引发的非特异性扩增依然存在,并且会对目标基因的扩增效率产生影响。
在前处理问题上,高通量的前处理装置已经逐步用于食品样品的快速检测,例如基于纳米级中空纤维膜的农兽药高通量萃取装置[53]。细菌核酸提取的高通量、自动化前处理方法和处理装置的研究也有不少报道,这有助于进一步缩短等温扩增的检测时间,减少人工操作步骤,是等温扩增规模化检测的重要基础[54]。
在检测通量问题上,伴随着等温扩增芯片化的研究趋势(图7),等温扩增的体系得以不断缩小,单位面积可容纳的独立反应个数得到增加,这为解决检测通量问题提供了新的角度。Zhou等[55]设计了基于离心作用的LAMP微流控芯片,通过在圆盘形平面上构建独立反应通道来增加通量,可以同时检测10种常见的食源性致病菌。
4 结 论
综上所述,虽然等温扩增已有将近20年的历史,但因其准确性高、检测用时短、便携易操作的特点。目前依然作为快速检测和在线检测的主要手段被广泛应用于食源性致病微生物的检验检疫中。以LAMP和NEAR为代表的商业化现场检测产品的成功,也证明了等温扩增在微生物现场检测中的价值。等温扩增的相关研究持续受到较高的关注,其技术缺陷也正在被逐步克服,芯片化、高通量的等温扩增方法及其装置正在逐渐成为热点,随着技术体系的不断完善,其应用前景将会更加广阔。
