膦催化的吡咯烷-螺吲哚酮类化合物的合成
2019-06-04畅志鑫韩小瑜
畅志鑫,袁 征,韩小瑜,2
(1.浙江科技学院 生物与化学工程学院,杭州 310023;2.浙江省农产品化学与生物加工技术重点实验室,杭州 310023)
利用亲核性叔膦进攻活化的体系产生两性离子中间体,进而与各种亲电、亲核试剂反应构建C—C、C—N、C—O、C—S键及相应的环状链状化合物的方法,是近年来的研究热点[1-3]。三价叔膦具有强亲核性而碱性较弱,兼具良好的进攻性和离去性能,可有效稳定α-位负电荷,利于反应中间体的存在转化,易于接受β-位负电荷进攻再生叔膦,利于实现催化反应。正是由于膦的上述特点,使得n-π*相互作用下的含膦两性中间体具有发生多种反应的可能,同时,膦亲核性可调(变换膦上取代基)和中间体可规划的特点,给人们设计成系列的新型反应提供了良好的研究平台。在相关反应方法不断系统化涌现的同时,实现此类反应多元有机反应的应用也相继报道,尤其是膦催化的环加成反应,如[3+2][4]、[4+2][5-6]和[4+1][7-8]等环化反应,近年来得到了迅速的发展,许多反应已应用于天然产物和生物活性分子的合成当中[9]。
吡咯烷-螺吲哚酮类化合物是一类具有重要生物活性和结构特性的天然生物碱的核心结构,对产生生理活性有至关重要的作用[10]。吡咯烷-螺吲哚酮类生物碱最初是从夹竹桃科和茜草属植物中提取获得的[11]。这类化合物通常由色胺衍生而来,其核心结构由吲哚酮和-3位上的吡咯烷取代基共同组成,而该结构单元上不同的取代基又衍生出种类丰富的天然产物。由于其结构的特殊性,很多含有该结构单元的化合物表现出重要的生理活性。比如:天然产物Chitosenine对小鼠神经传导具有短暂的抑制作用[12];生物碱Spirotryprostatin A和B通过抑制微管蛋白聚合,阻断细胞周期,可以显著抑制肿瘤细胞的活性[13];而生物碱Srychnofoline也可以阻断不同肿瘤细胞的分裂[14]。
1 试验部分
1.1 材料和仪器
1.1.1 材 料
靛红、苄基溴、氢化钠、肼基甲酸叔丁酯、膦催化剂等药品,溶剂甲苯、二甲苯、四氢呋喃、乙醚、二氯甲烷、氯仿、乙酸乙酯、石油醚等均为分析纯,均购自上海安耐吉有限公司。
靛红衍生的Boc-亚胺和联烯酸酯根据文献[15-16]制备。
1.1.2 仪 器
电子分析天平BSA224S(德国赛多利斯集团);核磁共振仪AV500(德国布鲁克公司),熔点测定仪JH60(上海佳航仪器公司)。
1.2 试验方法
1.2.1 靛红衍生的Boc-亚胺制备
根据文献[15],以N-Bn靛红系列1-b为原料,可以通过图1方法得到酮亚胺1。
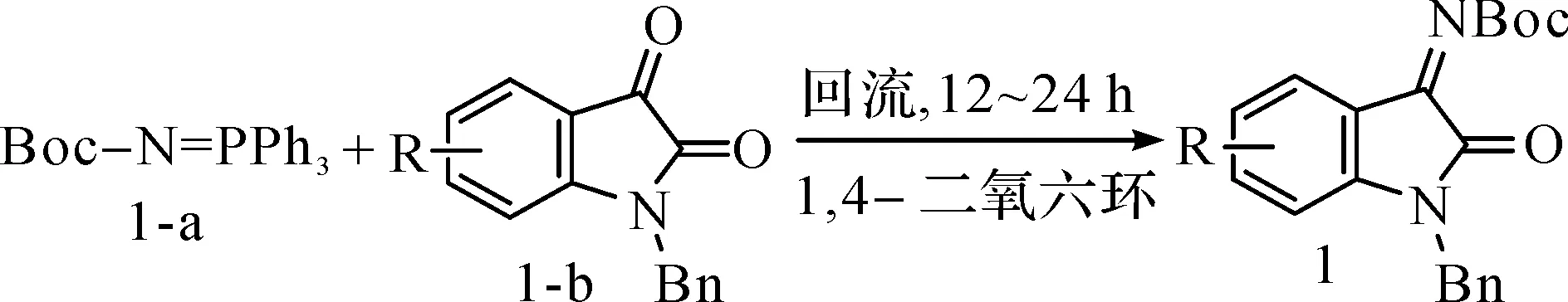
图1 靛红衍生的N-Boc亚胺的制备Fig.1 Preparation of isatin N-Boc ketimine
取烘干的25 mL圆底烧瓶,在氮气保护下,分别加入靛红1-b(10 mmol)和化合物1-a(11 mmol)。然后加入无水1,4-二氧六环(10 mL)溶剂,将混合物加热至回流,经TLC点板监测反应进行完全后,再将反应物冷却至室温。除去有机溶剂后,通过柱层析分离纯化得到酮亚胺1。
1.2.2 2,3-丁二烯酸苄酯制备
根据文献[16],以2-溴乙酸苄酯2-b为原料,可以通过图2方法得到2,3-丁二烯酸苄酯2。
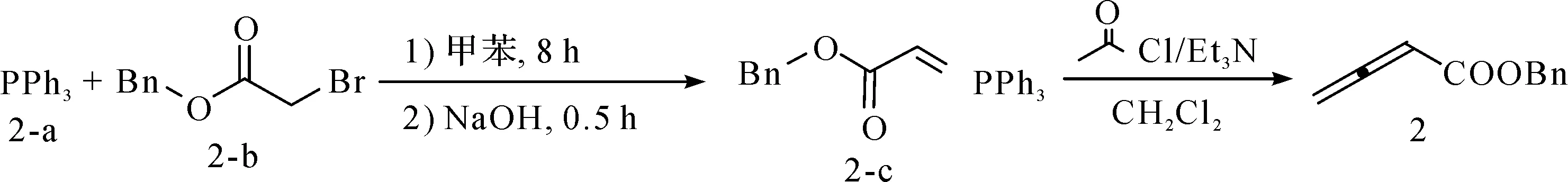
图2 2,3-丁二烯酸苄酯的制备Fig.2 Preparation of 2,3-butadienoic acid benzyl ester
称取PPh3(5.24 g,20 mmol)溶于甲苯(100 mL)溶剂中,随后加入溴乙酸苄酯(3.2 mL,3.58 g,20 mmol)在室温反应12 h,将反应生成的固体使用布氏漏斗过滤,并用石油醚洗涤、干燥,即得季鏻盐。之后将所得季鏻盐中加入2 mol/L的NaOH水溶液,并将该混合液室温(25 ℃)反应0.5 h,反应液用二氯甲烷(3×50 mL)进行萃取,饱和NaCl水溶液洗涤,最后用无水NaSO4干燥,除去有机溶剂,真空干燥得到固体化合物膦叶立德2-c(6.97 g,化学收率85%)。
称取上述膦叶立德2-c(4.10 g,10 mmol)溶于二氯甲烷溶液(20 mL)中,在0 ℃下加入Et3N(1.5 mL,11 mmol)及乙酰氯(0.78 mL,11 mmol),室温反应2 h。之后将反应液中的溶剂除去,并过滤除去反应体系中产生的固体,收集滤液,通过柱层析分离纯化得到2,3-丁二烯酸苄酯2(1.48 g,化学收率82%)。
1.2.3 膦催化联烯酸酯与酮亚胺的不对称[3+2]环加成反应
如图3所示,将化合物靛红亚胺1(0.1 mmol)溶于1 mL甲苯溶剂,加入联烯2(0.15 mmol),之后加入三苯基膦催化剂PPh3(0.1 mmol)。通过TLC点板监测反应并确认反应进行完全后,经柱层析分离纯化得到产物3。
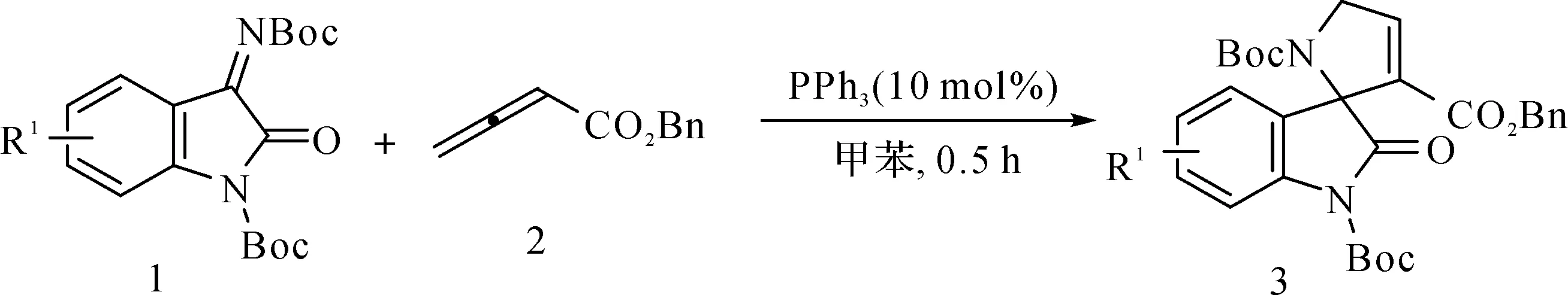
图3 三苯基膦催化[3+2]环加成反应Fig.3 Phosphine catalyzed [3+2] cyclization
2 结果和讨论
2.1 催化剂、溶剂及温度对[3+2]环加成反应的影响
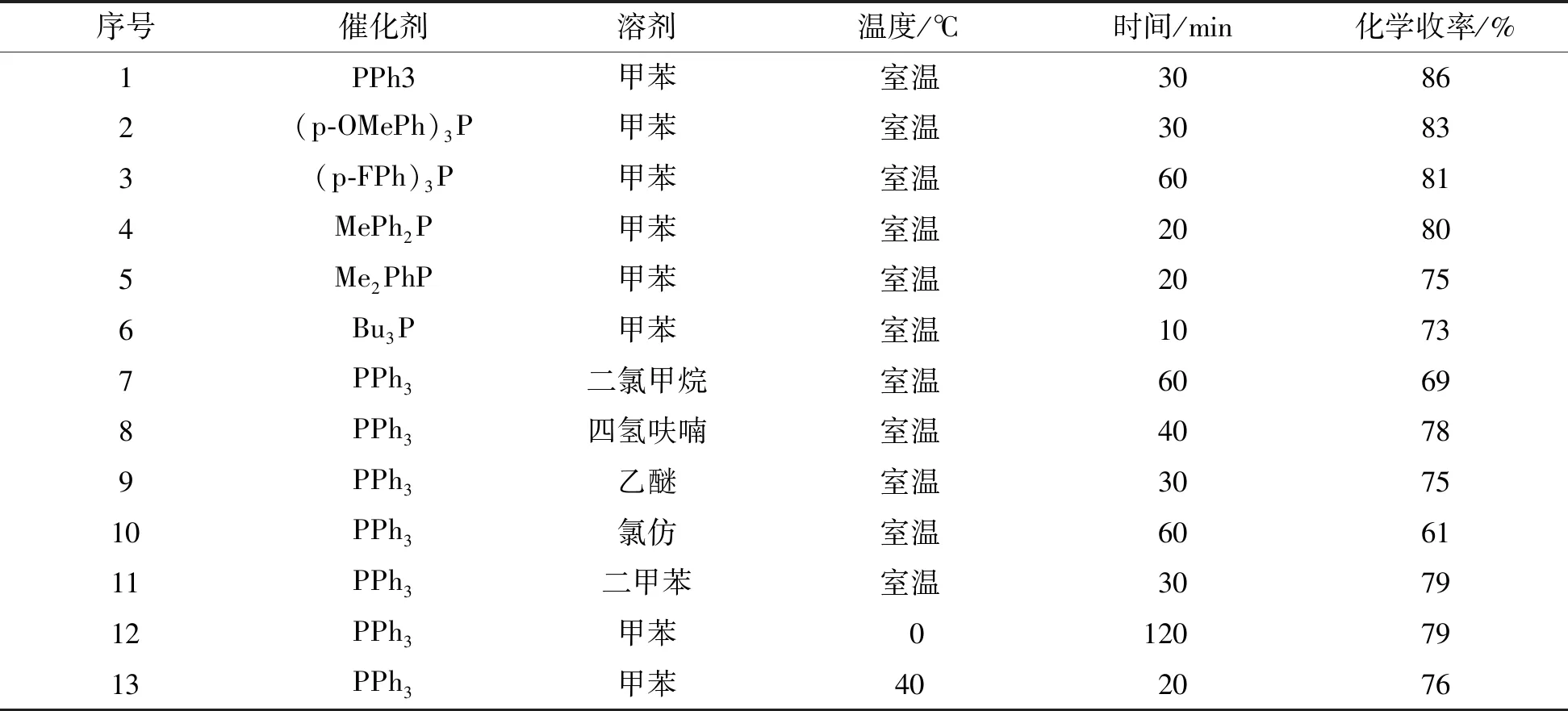
试验以靛红衍生的氮基-苄基取代的Boc-亚胺1a及苄基联烯酸酯2为反应底物,甲苯为反应溶剂,催化剂选取三价膦催化剂进行[3+2]环化反应研究(图4)。试验结果表明,几乎所有筛选的三价膦催化剂都表现出极高的催化活性(表1,序号1—5)。当该反应在甲苯中进行时,反应均可在1 h内反应完全。产物经柱层析分离提纯,可以以优良的化学收率得到目标产物。同时,三价膦催化剂的亲核性强弱对反应化学收率有一定的影响。表1中,随着膦催化剂亲核性的增强(PPh3 图4 三苯基膦催化[3+2]环加成反应条件优化Fig.4 Condition optimization of phosphine catalyzed [3+2] cyclization 此外,从试验结果来看,溶剂对反应有较大影响。我们选取了甲苯、二氯甲烷、氯仿、四氢呋喃、乙醚、二甲苯几种溶剂进行研究。苯类溶剂反应情况较好(表1,序号1、9),可以较高化学收率得到目标产物;醚类溶剂如乙醚和四氢呋喃也表现出较好的反应结果(表1,序号6—7),化学收率稍有降低;氯代甲烷类溶剂作为反应介质(表1,序号5、8)化学收率中等。同时,反应温度对环化反应也有影响,将该反应温度由室温降低到0 ℃时,反应时间延长至2 h,反应化学收率略有降低(79%)。升高温度至40 ℃时,反应速率增加,但反应化学收率同样降低,原因是有少量副产物产生。 表1 膦催化环化反应条件优化Table 1 Condition optimization of phosphine catalysis 以上试验结果表明,通过对靛红衍生的N-Boc-亚胺1a及苄基联烯酸酯2环加成反应的条件筛选优化,三苯基膦为最优反应催化剂,该反应以甲苯作为反应溶剂,室温,反应进行0.5 h,分离纯化得到的吡咯烷-螺吲哚酮3a的化学收率最高,因此被确定为该环化反应进行的最佳反应条件。 在以上最佳反应条件确定的基础上,我们进一步考察了该环加成反应对底物的适用性选择。试验结果表明(图5),靛红衍生的N-Boc亚胺反应物,当苯环不同位置取代时,底物对环化反应表现出优异的反应适用性。且亚胺的5,6,7-位连有供电子取代基如甲基、甲氧基,或者吸电子取代基如氟原子、氯原子吸电子基团时,反应都表现出极高的兼容性,所得目标产物的化学收率较高。另外,该反应底物亚胺的苯环上连有双取代基时,如产物3f所示,反应依然能表现出和其他底物相同的反应活性及反应效果。以上反应结果再次表明,我们所发现的三苯基膦催化的靛红衍生的氮基-苄基取代的Boc-亚胺1及苄基联烯酸酯2的环化反应的底物适用性较为广泛,是一种高效制备吡咯烷-螺吲哚酮类化合物的有机合成方法。所有分离产物都经核磁和高分辨质谱测试表征确认。 图5 三苯基膦催化[3+2]环加成反应的底物拓展Fig.5 Substrate extension of phosphine catalyzed [3+2] cyclization 3a:收率86%,白色固体;熔点141~142 ℃。1H NMR(500 MHz,氘化丙酮)δ7.87(d,J=8.3 Hz,1H),7.41(t,J=8.1 Hz,1H),7.35(t,J=2.2 Hz,1H),7.34-7.32(m,3H),7.27(dd,J=7.5,1.2 Hz,1H),7.20(t,J=7.5 Hz,1H),7.17(dd,J=6.6,3.0 Hz,2H),δ5.06(d,J=12.5 Hz,1H),4.99(d,J=12.6 Hz,1H),4.63(dd,J=18.5,2.1 Hz,1H),4.53(dd,J=18.5,2.3 Hz,1H),1.62(s,9H),1.12(s,9H);13C NMR(126 MHz,氘化丙酮)δ171.9,160.4,151.6,149.2,141.3,140.9,129.1,129.4,128.6,128.4,128.3,128.1,127.9,124.3,122.9,114.7,83.3,80.5,72.5,66.3,53.8,27.4,27.0;HRMS(ESI)m/z理论值C29H32N2NaO7[M+Na]+=543.212 5,实测值=543.210 2。 3b:收率84%,无色液体;1H NMR(500 MHz,氘化丙酮)δ7.74(d,J=8.3 Hz,1H),7.31-7.28(m,4H),7.21(dd,J=8.3,1.6 Hz,1H),7.15-7.14(m,2H),7.08(s,1H),5.05(d,J=12.5 Hz,1H),4.97(d,J=12.6 Hz,1H),4.60(dd,J=18.5,2.1 Hz,1H),4.50(dd,J=18.5,2.2 Hz,1H),2.31(s,3H),1.59(s,9H),1.11(s,9H);13C NMR(126 MHz,氘化丙酮)δ173.5,161.9,153.2,150.7,142.6,140.2,137.0,135.7,135.5,131.3,130.1,129.9,129.3,124.8,116.1,84.6,82.0,74.1,67.8,55.3,28.9,28.5,21.5;HRMS(ESI)m/z理论值C30H34N2NaO7[M+Na]+=557.225 8,实测值=557.227 6。 3c:收率79%,无色液体;1H NMR(500 MHz,氘化丙酮)δ7.84(d,J=1.9 Hz,1H),7.35(t,J=2.2 Hz,1H),7.33-7.29(m,4H),7.22(dd,J=8.0,1.9 Hz,1H),7.16(dd,J=6.6,2.9 Hz,2H),5.07(d,J=12.3 Hz,1H),4.95(d,J=12.2 Hz,1H),4.61(dd,J=18.5,2.2 Hz,1H),4.52(dd,J=18.5,2.3 Hz,1H),1.61(s,9H),1.13(s,9H);13C NMR(126 MHz,氘化丙酮)δ173.1,161.8,153.0,150.5,143.80,143.4,136.8,136.0,129.97,129.94,129.7,129.6,129.5,129.0,125.8,116.6,85.5,82.3,73.7,68.0,55.4,28.8,28.6;HRMS(ESI)m/z理论值C29H31ClN2NaO7[M+Na]+=577.171 2,实测值=577.172 4。 3d:收率88%,白色固体;熔点153~153.5 ℃。1H NMR(500 MHz,氘化丙酮)δ7.33(t,J=2.2 Hz,1H),7.29-7.27(m,3H),7.23-7.21(m,2H),7.16(dd,J=6.9,2.7 Hz,2H),7.11(d,J=8.6 Hz,1H),5.13(d,J=12.5 Hz,1H),4.98(d,J=12.4 Hz,1H),4.60(dd,J=18.5,2.2 Hz,1H),4.53(dd,J=18.5,2.3 Hz,1H),1.58(s,9H),1.16(s,9H);13C NMR(126 MHz,氘化丙酮)δ171.7,160.3,151.5,147.4,142.1,141.6,135.6,128.3,128.0,127.8,125.7,125.7,118.9,117.6,117.4,83.7,80.9,72.9,66.3,53.0,27.09,27.04;HRMS(ESI)m/z理论值C29H31FN2NaO7[M+Na]+=561.200 8,实测值=561.202 5。 3e:收率87%,无色液体;1H NMR(500 MHz,氘化丙酮)δ7.78(d,J=8.9 Hz,1H),7.31-7.29(m,4H),7.16(dd,J=6.6,2.9 Hz,2H),6.95(dd,J=8.9,2.8 Hz,1H),6.88(d,J=2.7 Hz,1H),5.05(d,J=12.5 Hz,1H),4.98(d,J=12.5 Hz,1H),4.61(dd,J=18.5,2.1 Hz,1H),4.50(dd,J=18.5,2.3 Hz,1H),3.77(s,3H),1.59(s,9H),1.13(s,9H);13C NMR(126 MHz,氘化丙酮)δ172.0,160.4,157.2,151.70,149.2,141.3,135.5,134.3,129.8,128.4,128.0,127.8,115.7,114.3,109.22,109.0,83.0,80.5,72.8,66.3,55.1,53.8,27.4,27.1;HRMS(ESI)m/z理论值C30H34N2NaO8[M+Na]+=573.220 7,实测值=573.222 7。 3f:收率82%,无色液体;1H NMR(500 MHz,氘化丙酮)δ7.27-7.25(m,4H),7.10(dd,J=6.5,3.1 Hz,2H),7.02(s,1H),6.87(s,1H),5.12(d,J=12.4 Hz,1H),4.93(d,J=12.5 Hz,1H),4.56(dd,J=18.5,2.2 Hz,1H),4.49(dd,J=18.6,2.3 Hz,1H),2.26(s,3H),2.19(s,3H),1.59(s,9H),1.13(s,9H);13C NMR(126 MHz,氘化丙酮)δ173.0,160.4,151.7,149.3,140.8,137.1,135.9,133.9,132.7,132.6,129.8,128.2,127.9,127.8,123.3,120.9,83.3,80.5,73.1,66.1,53.8,27.2,27.0,19.91,19.92;HRMS(ESI)m/z理论值C31H36N2NaO7[M+Na]+=571.241 5,实测值=571.243 0。 本文研究报道了三苯基膦催化苄基联烯酸酯与靛红亚胺的[3+2]环化反应。该反应条件温和,反应速度快,底物适用范围较为广泛,化学收率高,是一种高效制备吡咯烷-螺吲哚酮类化合物的有机合成方法,所有产物都经核磁和高分辨质谱表征确认。该膦催化环化反应的发现,进一步丰富了膦催化联烯酸酯与亚胺反应的类型,实现了膦催化酮亚胺与联烯酸酯的环化反应,为具有潜在生物活性化合物的合成提供了有效的方法参考。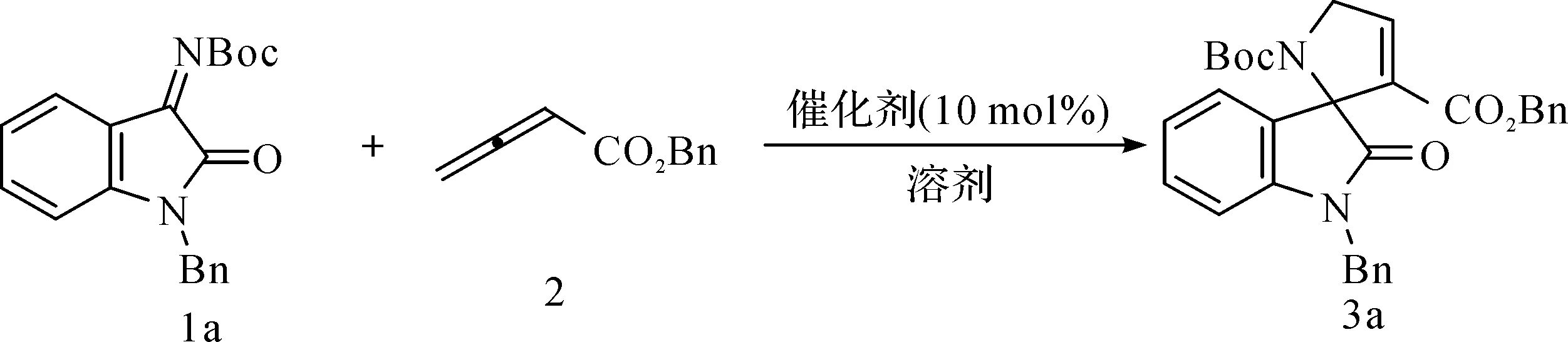
2.2 吲哚不同取代基对环化反应的影响

3 结 语
