过渡金属的能带结构与声子谱的第一性原理计算
2019-05-16陈春彩陈源福
陈春彩,陈源福
(闽南理工学院物理教研室,福建石狮362700)
过渡金属由于具有未填充的价层d轨道,导致他们的性质与其他元素有明显的差别。近年来过渡金属d电子对晶格相变机理的影响一直受到广泛的关注。过渡金属的声子谱对热力学性质具有非常重要的意义,Grabowski等采用PAW方法详细地研究了铜的声子谱及其对自由能、热容、热膨胀系数等热力学性质的影响,研究了这些热力学性质随温度的变化关系[1]。Alfè等人用分子动力学模拟的PAW方法计算了高温高压下液体铁的晶格结构和动力学性质[2]。卢志鹏等人通过采用基于密度泛函理论的第一性原理和准简谐晶格动力学方法对Ru的四种主要晶格结构的磁性和声子谱进行了系统的研究,得到各相结构的磁性基态及磁性稳定性范围[3]。胡翠娥等人基于经典的分子动力学模拟采用运用嵌入原子势模型研究了Mo的熔化曲线[4],认为只有考虑过热效应的两相模拟方法获得了钼的熔化温度数据才和动高压实验的结论一致,而单向模拟没有发生固固相变。赵凯[5]等人利用平面波赝势法同样计算了Mo在不同静水压下的声子色散关系及其熔化曲线,预测了620GPa处会发生BCC-HCP的结构相变,但是声子谱的计算结果在较高压力下与实验值误差较大,他认为研究相变路径还有待进一步深入的研究。马贺采用基于密度泛函理论的第一性原理运用CASTEP软件计算了金属Al的晶格常数、弹性系数、电子能带、态密度、声子谱、声子态密度以及相关热力学参数[6]。
文章利用第一性原理的密度泛函理论,运用abinit软件研究三种典型过渡金属Ni、Cu、Fe的晶格结构,电子能带、态密度,声子谱和声子态密度。分别计算它们的热容,熵,弹性模量,格林艾森参数和热膨胀系数等热力学函数并与实验结果比较。
1 计算方法
基于第一性原理的密度泛函理论(DFT),密度泛函微扰理论(DFPT),本文利用晶体结构计算程序包 abinit[7],采用广义梯度近似(GGA),Perdew-Burke-Ernzerhof(PBE)泛 函[8]和 Troullier-Martins(TM)赝势完成相应的第一性原理计算,计算时引入了电子的自旋极化。为了保证计算过程中能量得到有效收敛,分别优化计算得到平面波的截断能E-cut和k点的数量如表1所示。此外为了确保能量随晶格参数变化的连续性,特引入了光滑拖尾参数smearing和晶格参数的最大缩放比dilatmx。
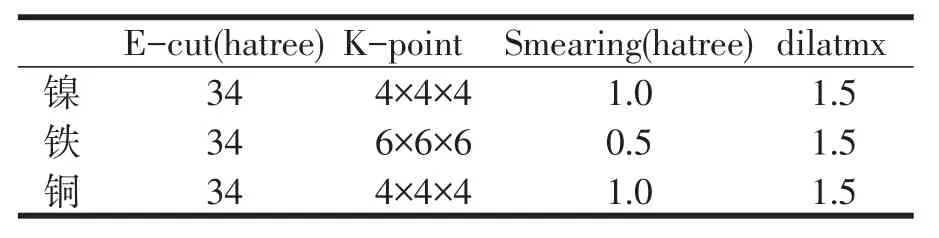
表1 优化参数
2 分析与讨论
2.1 晶格结构
金属Ni和Cu属于(FCC)面心立方结构,而Fe是(BCC)体心立方结构,分别优化计算得到Ni、Cu、Fe的晶胞参数、实验值如下表2所示。通过对比可知,偏差分别为1.3%、0.46%、3.9%。
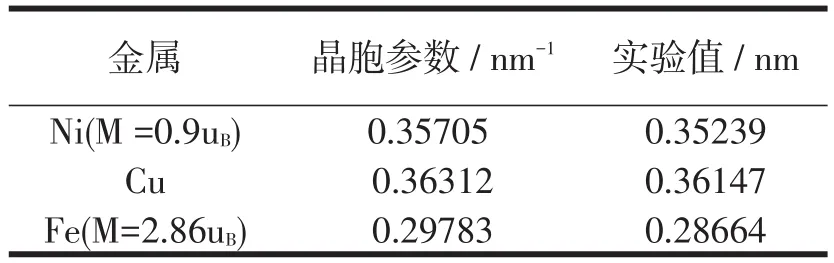
表2 Ni、Cu、Fe的晶胞参数与实验值
2.2 电子能带与态密度
镍属于铁磁质,宏观表现是有磁矩的,所以电子自旋向上和向下的态密度不是彻底对称的关系,图1代表镍自旋向下的电子能带和态密度,而图2代表自旋向上时的电子能带和态密度。在费米面(Fermi)处,有电子能带穿越费米面,形成较宽范围的导带,表现出明显的金属性。自旋向上电子能带在费米面处的态密度接近为零,而自旋向下电子态密度的峰值在Feimi面下约-1.57 eV处形成一个大的态密度峰,峰值是1.47 eV-1,这主要归功于L-XW区间的能带电子作出了贡献。由此电子能带图可知,当晶格遭遇热或外场的感化作用时,激发的电子最主要的还是由L-Г-X-K区间自旋向下的电子贡献。
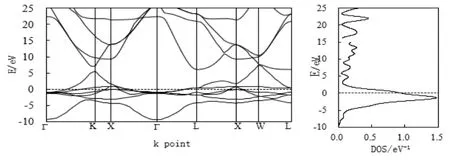
图1 Ni自旋向下电子能带与态密度
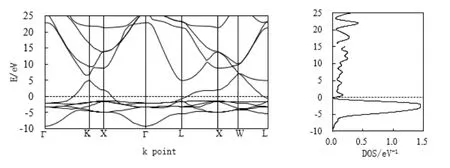
图2 Ni自旋向上电子能带与态密度
金属Cu是抗磁质,抗磁性物质的原子的磁矩应为零。图3是铜的电子能带结构和态密度图。对比发现铜的电子能带与态密度和金属镍自旋向上的电子能带和态密度的形状有非常相似的特性,大致的趋势相同,除了铜的态密度峰峰值偏小约0.64 eV-1。Cu和Ni虽然在元素周期表上是相邻的,也都属于过渡金属,但价层d轨道电子的排列方式完全不同,Cu的最外层电子排列是3d104s1,而Ni的外层电子排列是3d94s1。因为4s是宽带,3d是窄带,在费米面处Cu原子在4s价带以后的d轨道电子数是彻底填满的,因此3d轨道上的电子将不介入传导,而Ni的3d轨道电子数没有彻底填满,有3d轨道的电子会介入局域传导。
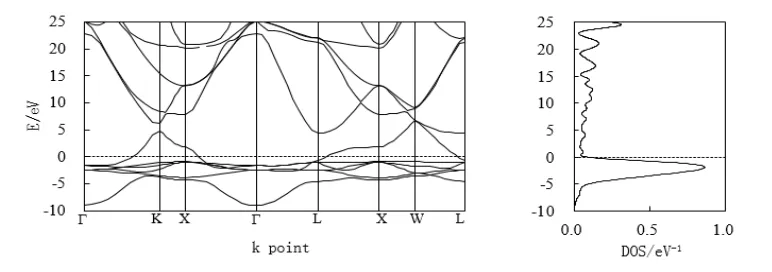
图3 Cu的电子能带与态密度
金属Fe是铁磁质物质,磁矩M=2.86 uB。图4、图5分别代表金属铁自旋向上和自旋向下电子的能带结构和态密度。在费米面能级处,有能带穿越费米面,构成导带的范畴较宽,体现出较强的金属性。自旋向上电子能带在费米面处的态密度相对很小,似乎可以不计。自旋向下电子态密度的峰值在Feimi面处构成两个较锋利的态密度峰。在费米面上2.02 eV有最大的态密度峰,峰值为0.73 eV-1;在费米面下-0.42 eV处,有第二个较大的态密度峰峰值为0.58 eV-1。主要由Γ-P-N-H区间自旋向下的d轨道电子和s轨道电子提供的贡献。

图4 Fe自旋向上电子能带与态密度
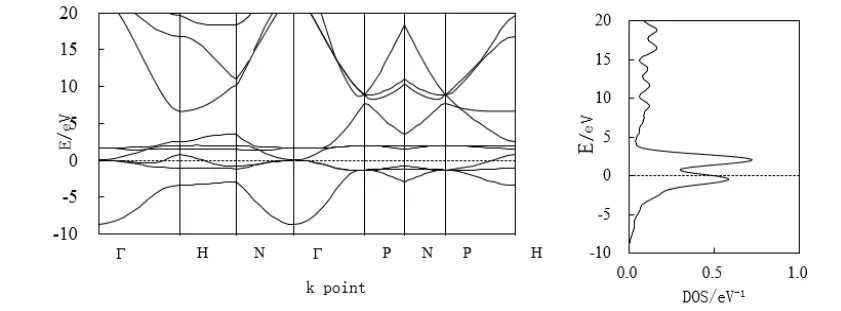
图5 Fe自旋向下电子能带与态密度
2.3 声子谱与态密度
应用密度泛函理论及其微扰理论,优化金属Ni、Cu、Fe的线性响应函数和晶格动力学矩阵,便可计算获得对应的声子谱。图6,图7,图8分别是金属Ni、Cu、Fe的声子能带结构及其态密度,实线代表本文的计算结果,点代表的是实验结果,虚线代表其他理论计算结果。由图可知,声子能带包含三个声学波带,即一个纵声学(LA)波带和两个横声学(TA)波带。通常情况下,LA的能量略高于TA的能量,而在对称性较高的Г点能量都为零。

图6 Ni的声子能带结构与态密度

图7 Cu的声子能带结构与态密度

图8 Fe的声子能带结构与态密度
由于过渡金属Ni和Cu在元素周期里是相邻的,都具有面心立方结构,所以声子谱和态密度外形存在高度的一致性,都是在Г点附近较大范围内,横声学波带是二重简并的,但在K、W点附近以及L-X之间这种二重简并就消失。横声学(TA)波带和纵声学(LA)波带分别构成了两个比较大的态密度峰,其中Ni的峰值分别为179 cm-1、256 cm-1,Cu的峰值分别为132 cm-1、238 cm-1。对过渡金属Fe,除横声学波带在Г-H、Г-P和P-H之间有二重简并外,在Г-N-H之间,N-P之间简并消逝,Fe的态密度峰相对Cu和Ni对比不明显,但计算获得的这三种过渡金属的声子能带变化规律总体与实验结果吻合较好。
图7 还给出了Grabowski等[1]采用PAW方法计算得到Cu的声子谱。本文的计算结果与PAW方法得到的结论相当,但在是声子能量较高处本文的结果明显优于PAW方法的计算结果。
2.4 热力学性质
应用声子态密度计算获得Ni、Cu、Fe在298.15K下的等体热容CV和熵S。应用晶格参数与晶格能量之间的变化关系计算获得体积模量B和格林艾森参数γ,同时还可计算得到体系的体胀系数α,分别与实验结果比较,所得误差值均列于表3。
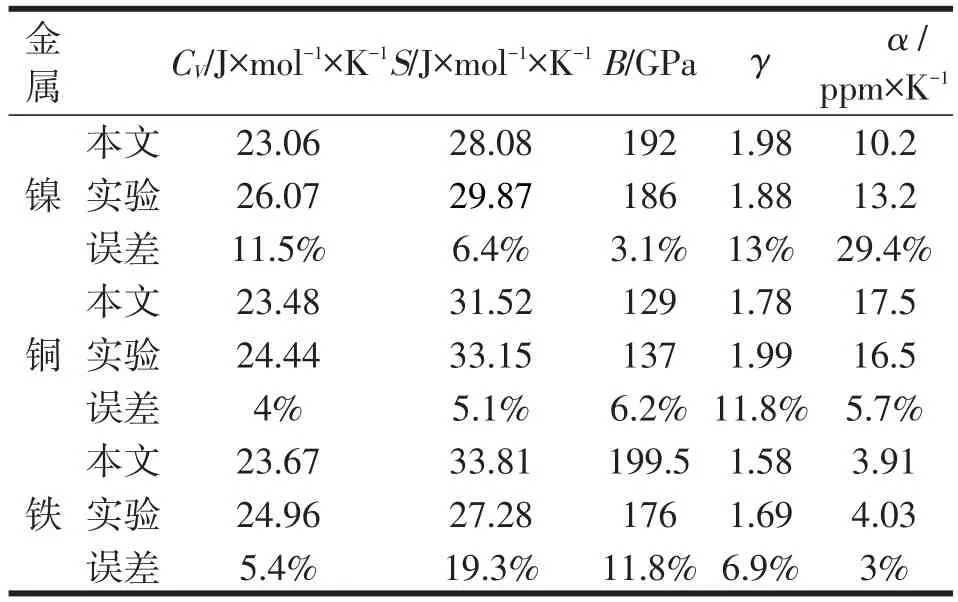
表3 Ni、Cu、Fe在298.15K下的热力学参数
3 结语
本文基于密度泛函理论,结合第一性原理应用abinitio软件计算了过渡金属Ni、Fe、Cu的电子结构、声子能带与热力学性质,得出如下结论:
1)通过对电子能带结构的剖析,得到数据表明镍和铁有磁矩,而铜没有磁矩。可是三种金属的电子能带在费米面处都存在相交,说明都具有显著的金属性。
2)面心立方过渡金属铜和镍就有类似的声子能带构造和态密度峰,而体心立方金属铁有3条声子谱曲线,态密度峰比铜和镍的小许多。在对称点高的Г点周围较大范围内,理论计算结果与实验结果吻合得非常好。
3)计算得到的热力学相关数据与实验结果吻合较好。
