外电场作用下CF3I分子结构特性及性质
2019-04-29李亚莎刘国成刘志鹏谢云龙
李亚莎, 刘国成, 刘志鹏, 谢云龙, 徐 程
(三峡大学电子与新能源学院,宜昌443002)
1 引 言
随着电力行业的飞速发展,SF6由于良好的绝缘能力和灭弧性能被广泛用于气体绝缘组合电器(GIS)、气体绝缘断路器(GIB)等电力设备中[1-3],但SF6的温室效应潜在值(GWP)为CO2的23900倍[4,5],对环境有极强的破坏作用. CF3I作为一种潜在的SF6替代性气体是近年来研究的热点,CF3I是一种无色、无味、无毒、具有较强电负性的气体,它的温室效应潜在值(GWP)小于5,在大气中的寿命只有0.005 a[4,5]. 目前,对于CF3I及其混合气体的绝缘性能进行了大量的研究. 文献[6]研究得出CF3I在雷电冲击电压下的击穿电压约为纯SF6气体的1.2倍. 文献[7]通过能量为0-40 eV的电子碰撞CF3I气体,测出了在此电子能量范围内CF3I的电子亲和势为0.32±0.2 eV. 文献[8]通过近似求解Boltzmann方程,求出在一个大气压下50%SF6/50%CF3I混合气体的击穿场强,得出了在温度为1800℃以上时,其击穿场强高于纯SF6气体临界击穿场强的结论. 文献[9,10]基于密度泛函理论(DFT)研究了CF3I在微水、微氧环境中的分解产物微观形成机理,并且采用过渡态搜索计算了CF3I反应物与生成物之间反应的活化能及反应热.
受外电场作用,分子内电子在外电场作用下会吸收一定的能量,由能量较低的基态跃迁至能量较高的激发态,分子活性增大,外电子碰撞下容易发生分子键断裂,进而形成自由基,易与其他物质发生化学反应,其物理及化学性质变化极大[11]. 文献[12]采用密度泛函理论的方法研究了在外加电场-0.04~0.04 a.u.下CHCl3的分子结构与激发特性,随着电场的增加,分子结构发生形变,分子的能隙先增大后减小,分子反应活性增加. 文献[13, 14]采用密度泛函理论方法分别研究了SnS和CaS分子在外电场作用下分子结构及其激发特性,并且分析了此分子前9个激发态的激发能、振子强度和吸收波长.
CF3I在外电场作用下结构及性质研究还鲜有报道,因此对在外加电场下CF3I分子结构及激发态进行研究很有必要. 本文采用HF方法在3-21g基组下研究了CF3I分子的基态分子键长、偶极距、前线轨道等参数,并且在此基础上采用杂化CIS/3-21g方法计算外电场对于CF3I分子前9个激发态的激发能、吸收峰、振子强度.
2 理论计算
外加电场下分子的哈密顿量H可以用下式来表示[15]:
H=H0+Hint
(1)
H为分子在未加电场时的哈密量;Hint为外电场与分子体系相互作用时的哈密顿量. 在偶极近似的情况下,分子体系与外电场相互作用能[18]如下式子:
Hint=-μ·F
(2)
μ为电偶极距,F为外电场作用力.
对CF3I分子沿X轴方向加电场强度为0~0.009 a.u. (约为0~4.62106kV/m)范围电偶极化电场,这里加的电场较强,是对CF3I分子结构的作用,大于CF3I气体的击穿场强(在0.1 MPa气压,间隙为10 mm时,CF3I击穿场强约为10.5 kV/mm).

图1 CF3I分子结构模型Fig.1 Structure of CF3I
3 结果与讨论
在3-21g(碘原子超出6-31g基组级以上基组适用范围,因此选用3-21g基组)基组的情况下,分别选用HF、LSDA、BPV86、B3LYP、B3PW91方法对CF3I分子优化,对比计算结果,采用HF方法计算的键长与实验键长结果最为接近,C-F键长为0.13406 nm,实验值为0.1347 nm[17],C-I键长为0.21769 nm,实验值为0.2201 nm[17],验证了采用HF/3-21g方法来进行CF3I分子优化计算的准确性. 图1为Gaussian View 5.0上建立的CF3I模型,蓝色为F原子,灰色为C原子,紫红色为I原子.
3.1 CF3I在外加电场下键长的变化
在外加电场时采用HF/3-21g方法优化基态CF3I分子结构的键长数据如表1所示,由表1可以看出F2-C1、F3-C1和F4-C1键长随着电场的增加而减小,分子键能增加;I5-C1键长随着电场强度增加而增加,分子键能减小,更容易断裂,与文献[18]研究得出CF3I在电场作用下,带电粒子与CF3I发生碰撞,C-I极易断裂的结论一致. 分子几何参数的变化可以用电荷转移引起分子内电场变化来定性解释[13,21],上述键长的变化表明,随着外电场沿图1所示X轴方向增加,F2-C1、F3-C1和F4-C1局部电场强度增加,键长减小,I5-C1局部电场减小,键长增加. 键长的变化也表明在外电场作用下,CF3I分子结构发生变形.
表1 外加电场下CF3I分子键长变化
Tab.1 Bond length of CF3I molecule under different applied electric field
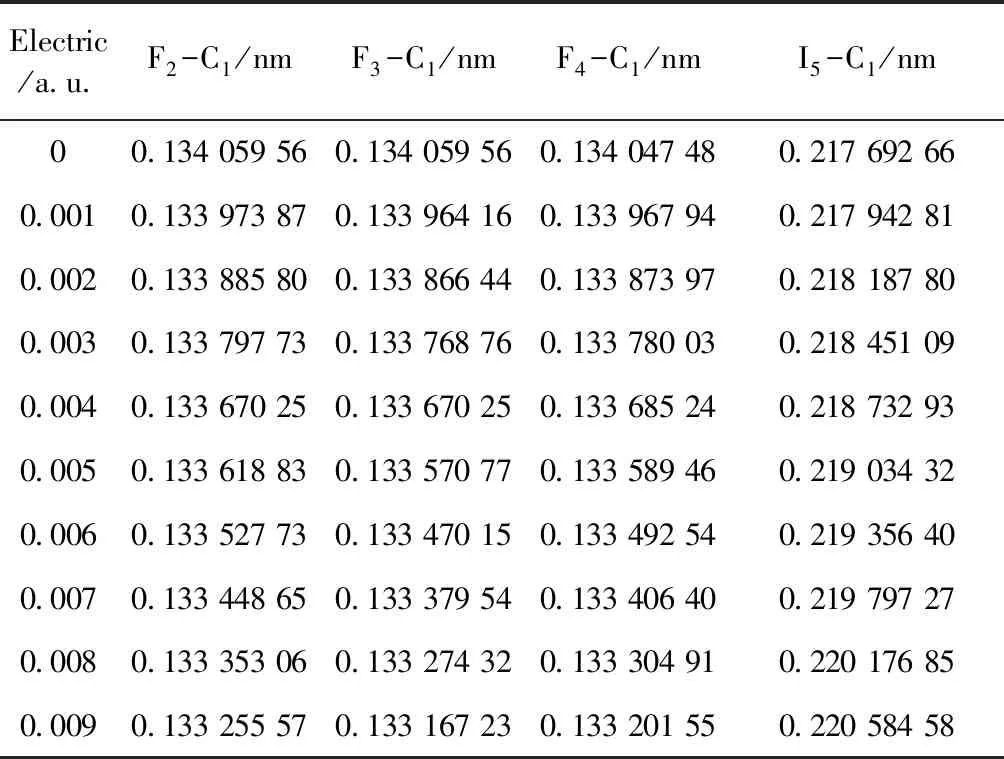
Electric/a.u.F2-C1/nmF3-C1/nmF4-C1/nmI5-C1/nm00.134 059 560.134 059 560.134 047 480.217 692 660.0010.133 973 870.133 964 160.133 967 940.217 942 810.0020.133 885 800.133 866 440.133 873 970.218 187 800.0030.133 797 730.133 768 760.133 780 030.218 451 090.0040.133 670 250.133 670 250.133 685 240.218 732 930.0050.133 618 830.133 570 770.133 589 460.219 034 320.0060.133 527 730.133 470 150.133 492 540.219 356 400.0070.133 448 650.133 379 540.133 406 400.219 797 270.0080.133 353 060.133 274 320.133 304 910.220 176 850.0090.133 255 570.133 167 230.133 201 550.220 584 58
3.2 外电场作用下CF3I Mulliken电荷布居分布、红外光谱分析及分子偶极距
表2为基态CF3I分子在外加电场的作用下优化得到的Mulliken电荷布居分布,由表2可以看出在外电场的作用下,F2、F3、F4和C1的电荷随着外加电场强度的增加而增加,I5的电荷量随着外加电场强度的增加减少. 这说明在外电场的作用下,F2、F3、F4和C1电负性减小,I5电负性增加,分子内电子远离F2、F3、F4和C1,导致F2、F3、F4和C1上电荷增大,分子内电子偏向I5,致分子内部的I5电荷减小.
下图2为CF3I分子的红外光谱图,在未加外电场时,分子有四个特征吸收峰,波数分别是547.89 cm-1、755.48 cm-1、1186.51 cm-1和1386.26 cm-1. 其中吸收峰1(547.89cm-1)对应C1-I5伸缩振动吸收峰,与文献[19]提供C1-I5伸缩振动吸收峰波数523 cm-1基本相符,特征吸收峰2(755.48 cm-1)对应CF3·(CF3基团)弯曲振动,与实验值CF3·弯曲振动吸收峰为743 cm-1基本相符[20],特征吸收峰3(波数1186.51 cm-1)和特征吸收峰4(波数1386.26 cm-1)对应CF3·的对称与非对称伸缩振动,与文献[19]提供CF3·的对称与非对称伸缩振动值1148 cm-1,1278 cm-1基本相符,验证了计算的正确性. 施加外电场后,分子特征吸收峰2在电场为0.003 a.u.时,波数为755.88 cm-1,发生蓝移现象,之后在电场为0.006 a.u.和0.009 a.u.时,波数分别是755.77 cm-1和754.55 cm-1,又出现红移现象,表明随着电场的增强CF3·基团振动先是剧烈后又缓和;特征吸收峰3在电场为0.003 a.u.、0.006 a.u.及0.009 a.u.时,波数分别为1157.98 cm-1、1173.07 cm-1和1186.51 cm-1,随着电场增强出现红移现象,表明对应CF3·非对称振动更加缓和;在电场0.003 a.u.、0.006 a.u.及0.009 a.u.时,特征吸收峰1波数分别为548.22 cm-1、549 cm-1和549.27 cm-1,特征吸收峰4波数分别为1399.29 cm-1、1412.73 cm-1和1246.21 cm-1, 1、4特征吸收峰在电场作用下蓝移现象明显,表明对应的CF3·弯曲振动和CF3·的对称伸缩振动更加剧烈. 外电场对分子的内部基团振动影响明显,影响了分子红外吸收光谱.
CF3I分子在外电场作用下的偶极距变化如图3所示,根据图3可知分子的偶极距随着正X轴方向电场强度增加(电场范围0~0.009 a.u.)逐渐减小,表明对图1所示的CF3I分子在沿X轴正方向加电场,分子偶极距减小,分子趋于非极性分子.
3.3 CF3I分子前线轨道、能隙及总能量变化
分子前线轨道分为最高占据轨道(HOMO)与最低未占据轨道(LUMO),HOMO轨道上能量反映分子失去电子的能力,HOMO轨道的能量越高,该分子越容易失去电子;LUMO轨道反映分子得到电子的能力,在数值上与分子的电子亲和势相当,LUMO轨道的能量越低越容易得到电子[13]. 能隙表示分子中电子从HOMO轨道跃迁到LUMO轨道的难易程度,能够表征分子的活性,能隙越小,分子活性越大. 能隙计算公式如下:
Eg=ELUMO-EHOMO
(3)
Eg为分子的能隙;EHOMO为分子的HOMO轨道能量;ELUMO为分子的LUMO轨道能量.
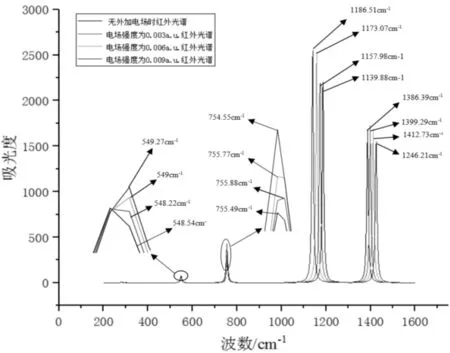
图2 CF3I在外电场下的红外光谱分析Fig. 2 Infrared Spectrum Analysis under different Electric Field

图3 外电场作用下CF3I偶极距变化Fig. 3 Change of CF3I dipole moment under different electric field
根据表3可以知道HOMO轨道能量在0~0.006 a.u.之间逐渐减少,在0.007~0.009 a.u.之间微弱波动,LUMO轨道能量随着电场的增加逐渐减少,由图4 可知分子的能隙随着电场强度的增加逐渐减小,这表明,分子内电子从HOMO轨道跃迁到LUMO轨道所需要的能量减少,电子易从HOMO轨道激发至LUMO轨道,HOMO轨道形成空穴[21].
根据图5分子总能量的变化可以看出,CF3I分子总能量随着外电场的增加而增加,这是因为在外电场的作用下,电子远离F2、F3、F4,且靠近I5,致使F2、F3、F4电荷布居数(绝对值)减小,且I5的电荷布居数减小,从而使得体系的哈密顿量中的势能减小,进而导致分子的总能量增大[13].
表2 外电场作用下CF3I电荷布居分布及原子间电荷布居数差
Table 2 CF3I Charge Distribution and Intersatomic Charge Distribution under different Electric Field
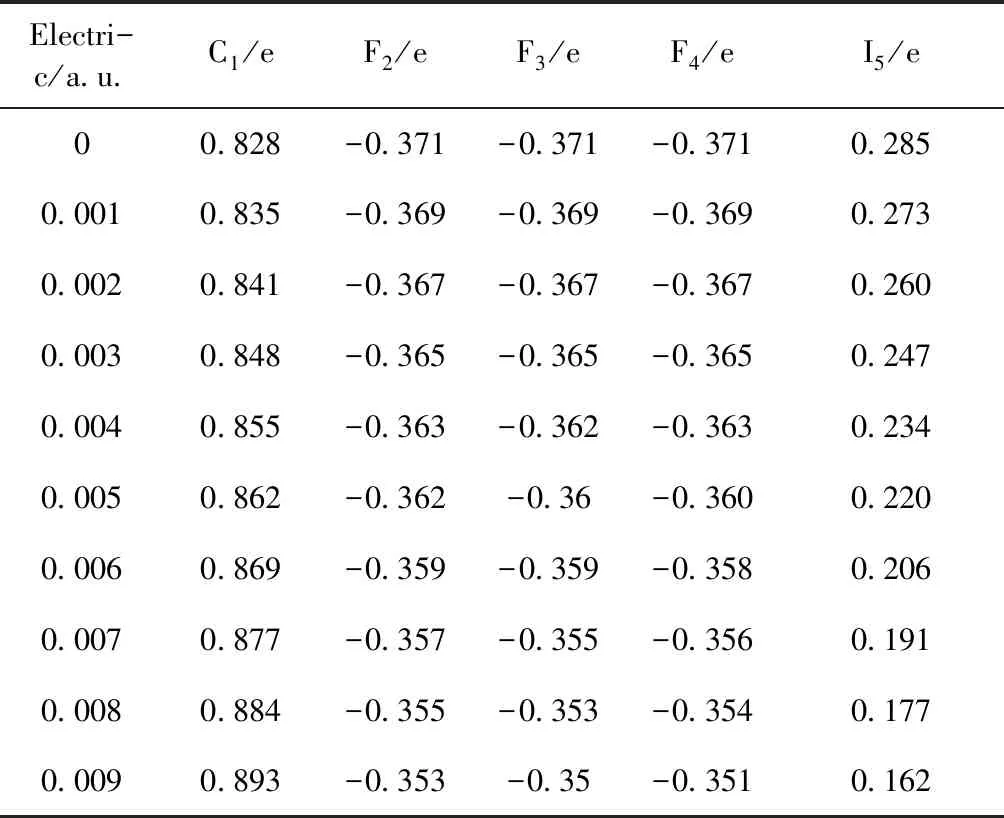
Electri-c/a.u.C1/eF2/eF3/eF4/eI5/e00.828-0.371-0.371-0.3710.2850.0010.835-0.369-0.369-0.3690.2730.0020.841-0.367-0.367-0.3670.2600.0030.848-0.365-0.365-0.3650.2470.0040.855-0.363-0.362-0.3630.2340.0050.862-0.362-0.36-0.3600.2200.0060.869-0.359-0.359-0.3580.2060.0070.877-0.357-0.355-0.3560.1910.0080.884-0.355-0.353-0.3540.1770.0090.893-0.353-0.35-0.3510.162
3.4 外电场对CF3I分子的激发态的影响
在优化得到CF3I分子基态结构的基础上,采用杂化CIS方法在3-21g的基组上对CF3I分子进行激发态计算,得到分子的前9个激发态激发能、吸收峰和振子强度. 激发能反映分子从基态到激发态所需能量. 表4为CF3I分子在外电场作用下激发能. 由表4数据可以看出,CF3I分子第1、2和9激发能都是随着电场强度增大,呈现先增大后减小趋势,且1、2激发能在0.005 a.u.电场强度时,分子激发能最大,激发能分别是2.5440 eV和2.5452 eV,第9激发态在0.004 a.u.时激发能最大,激发能为13.2452 eV;第3、4、5、6、7和8激发态随着电场强度的增加,激发能逐渐减少,说明对图1所示CF3I分子沿X轴方向对CF3I分子施加外电场时,随着电场强度的增加,第3、4、5、6、7和8激发态越来越容易被激发. 从激发能大小的角度分析,第1、2激发态的激发能相比于其他激发态小很多,激发所需的能量较少,激发相对容易. 激发能的变化可以由分子轨道能级的升降以及电场改变了分子轨道电子跃迁状态共同解释[21]. 如分子在无外加电场时,分子从基态激发至激发态8时,激发能为11.1869 eV(约为1079.37 kJ/mol),跃迁轨道是HOMO36LUMO44;在外加电场为0.005 a.u.时,激发能为11.1019 eV(约为1071.17 kJ/mol),电子跃迁轨道为HOMO37LUMO44跃轨道.
表3 CF3I分子在外加电场下的分子前线轨道能量数据
Table 3 Molecular frontier orbital energy data of CF3I molecules under different applied electric field
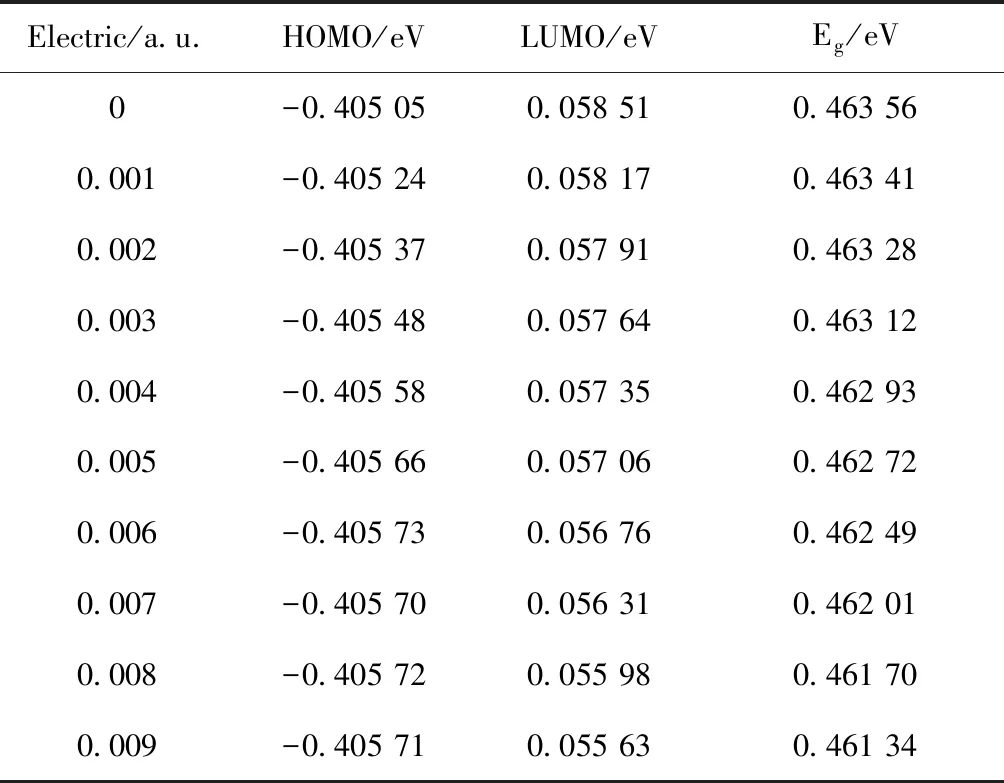
Electric/a.u.HOMO/eVLUMO/eVEg/eV0-0.405 050.058 510.463 560.001-0.405 240.058 170.463 410.002-0.405 370.057 910.463 280.003-0.405 480.057 640.463 120.004-0.405 580.057 350.462 930.005-0.405 660.057 060.462 720.006-0.405 730.056 760.462 490.007-0.405 700.056 310.462 010.008-0.405 720.055 980.461 700.009-0.405 710.055 630.461 34
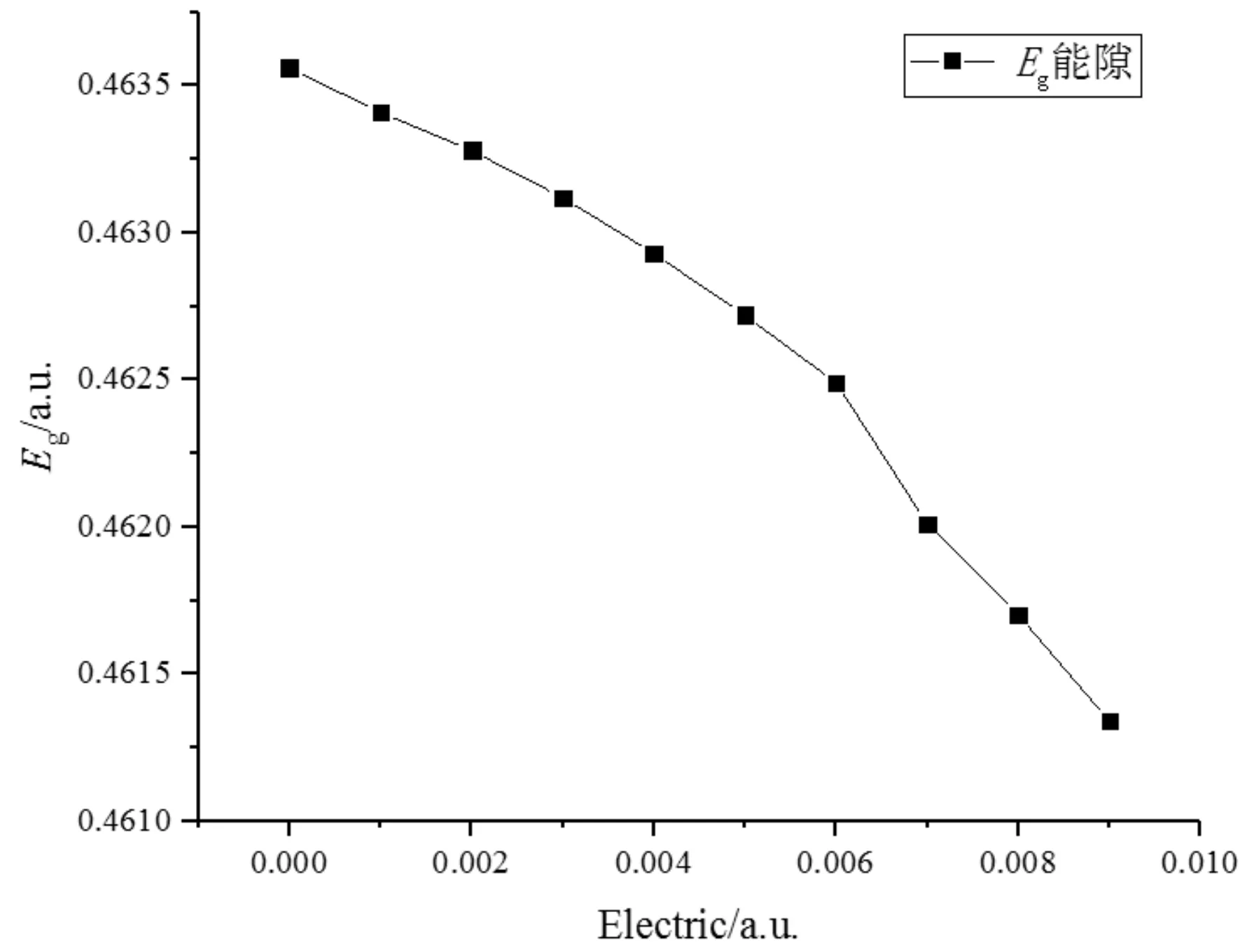
图4 外电场下CF3I分子能隙变化Fig. 4 Energy gap of CF3I molecule under different electric field
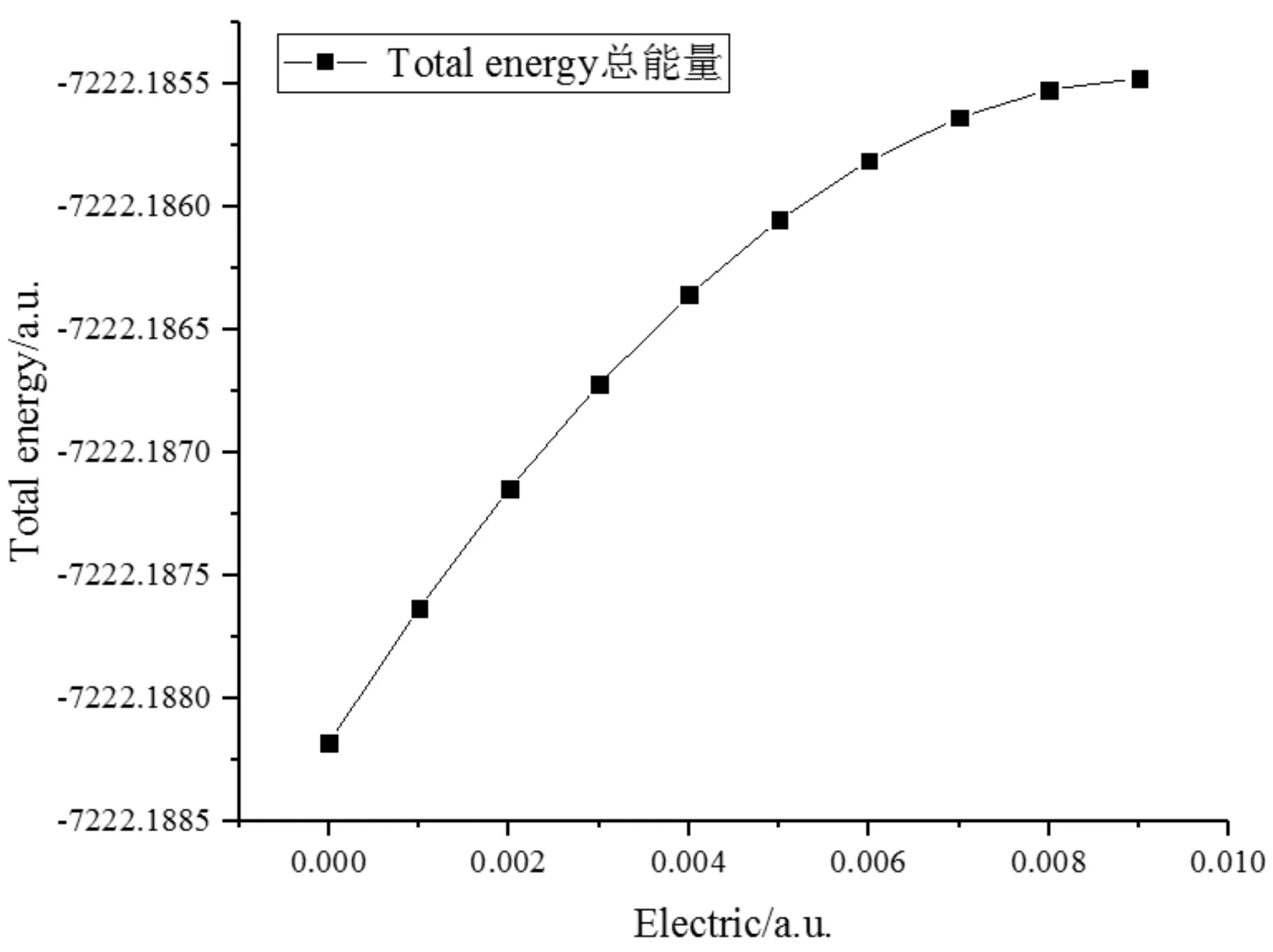
图5 外电场下CF3I分子总能量变化Fig. 5 Changes in total energy of CF3I molecule under different electric field
CF3I在外电场作用下的激发态波长数据如表5所示,由此可以看出整个光谱的波长范围在90~500 nm之间,属于紫外光谱和可见光谱的范围. 从表5还可以看出分子在外电场的作用波长发生了变化,第1、2和9激发态吸收波长先减小后增大,且1,、2激发在电场强度为0.005 a.u.时,吸收波长最短,波长分别为487.37 nm和487.13 nm,第9激发态在电场强度为0.004 a.u.时,吸收波长最短,波长为93.61 nm,第3、4、5、6、7和8激发态波长随着电场强度的增加而增加. 这同样可由电子跃迁状态的改变解释,分析上述激发态9,在外加电场存在时,激发态9电子跃迁轨道为HOMO34LUMO44,跃迁波长为94.5 nm;在外加电场强度为0.005 a.u.时,电子跃迁轨道为 HOMO32LUMO44,波长为93.63 nm. 由此可知吸收波长受外电场的作用影响较大,可以由外电场强度改变CF3I的电子跃迁发光光谱波长.
表6为CF3I分子在外电场作用下的振子强度数据,振子强度反映了分子各个激发态的激发能力,振子强度越大,该激发态就越容易吸收相应频率的能量从基态跃迁至激发态. 根据表6的数据可以看出,在0~0.009 a.u.电场强度范围内,第1,、2、4、5、6激发态的振子强度为零,属于禁阻跃迁,激发态3振子强度的数值最大,激发能力最高,激发态3和9振子强度随着电场强度的增加而减小,激发能力逐渐减小,第7、8激发态的振子强度随着电场的增加而增加,这表明分子的第7、8激发态激发的能力相对增加. 表明外电场存在时, CF3I分子激发态跃迁的难易程度不同,可利用外电场能控制材料的发光强度[21].
综上所述可以看出,外电场影响了分子的激发能、波长和振子强度,且各个激发态的激发能、吸收波长和振子强度受电场的影响规律不完全相同.

表4 外电场作用下CF3I分子激发能
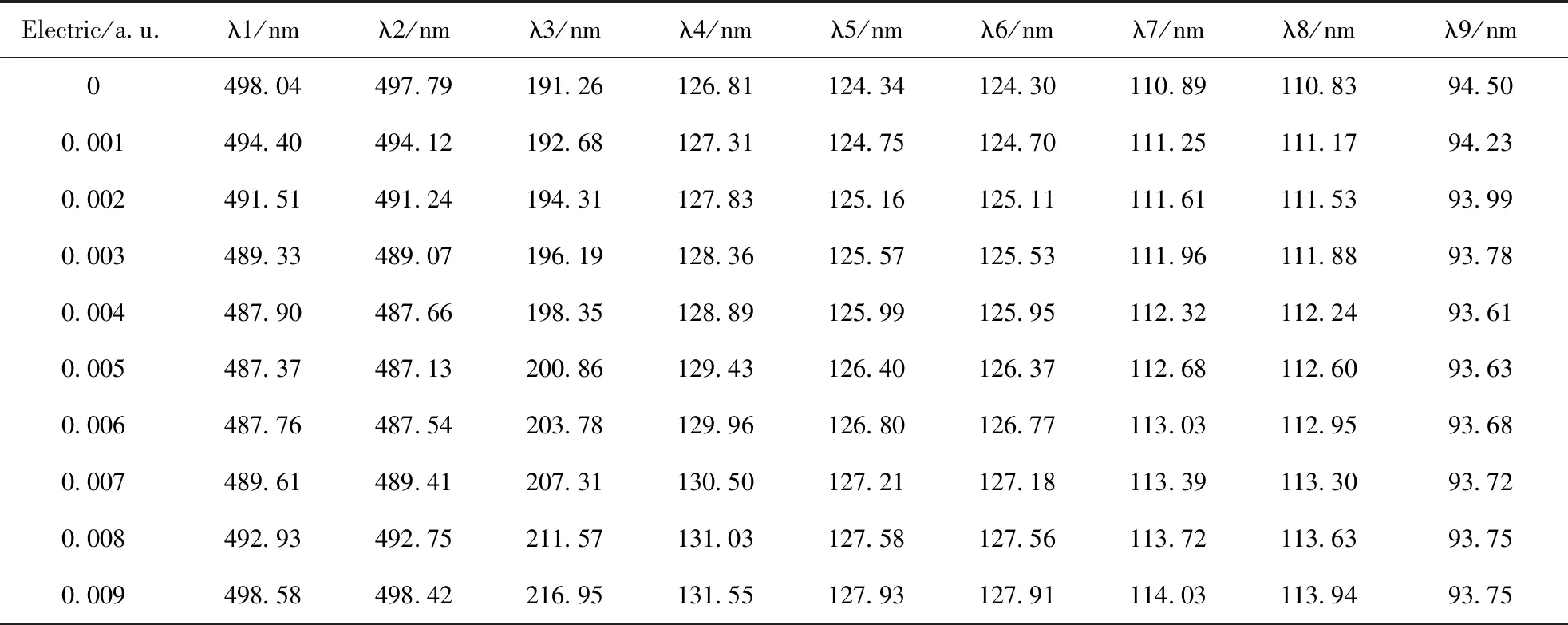
表5 外电场作用下CF3I分子吸收波长
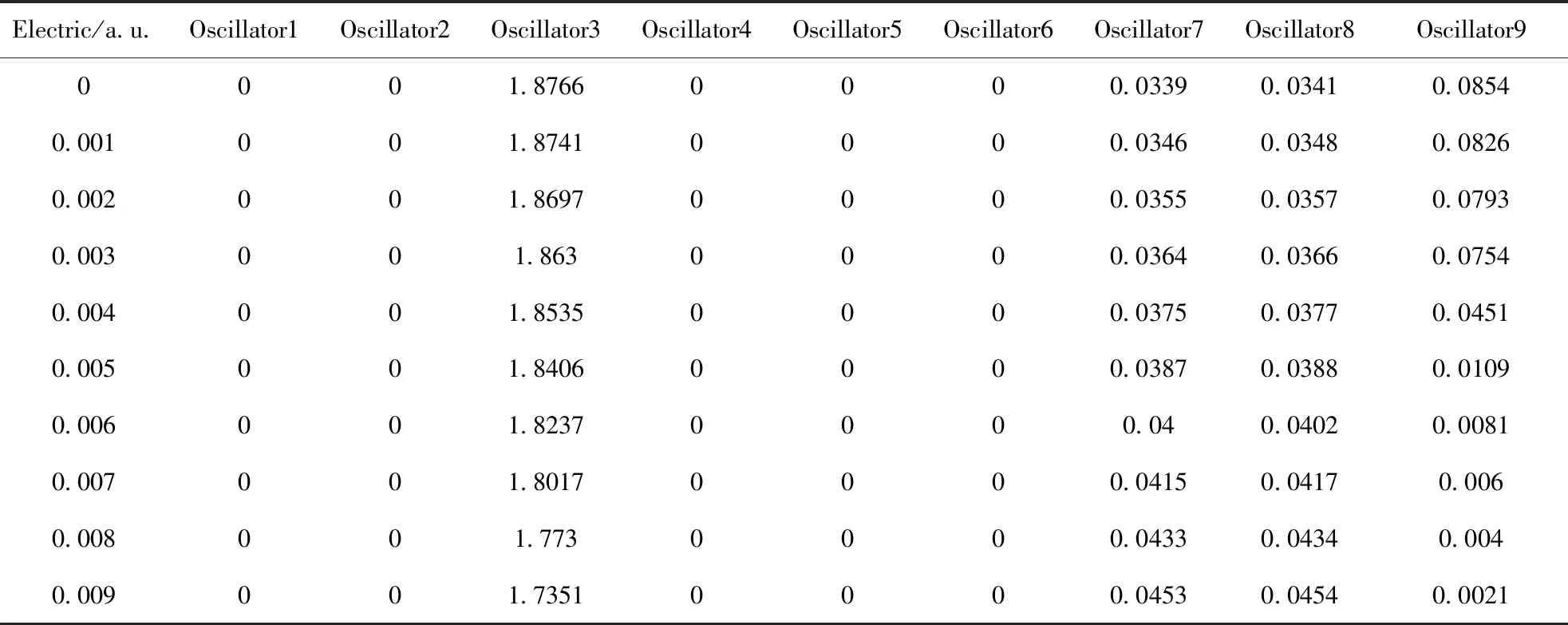
表6 外电场作用下CF3I分子振子强度
4 结 论
采用HF/3-21g方法优化了CF3I分子的基态结构,分析了分子的键长、偶极距、分子前线轨道、能隙、谐振频率和电荷布居分布;在此基础上还采用杂化CIS/3-21g方法计算了分子的前9个激发态,讨论了激发态的激发能、吸收光谱和振子强度. 得出如下结论:
1)分子的F2-C1、F3-C1和F4-C1键长随着电场强度的增加而减小,I5-C1键长随着电场强度的增加而增大,分子发生形变;分子内I5的电荷减小,其它原子的电荷逐渐增加,分子上电荷布居数逐渐由I5转向其它原子上.
2)CF3I分子的偶极距随着X轴正向电场强度增大而减小,分子逐渐趋于无极性分子,偶极距随着X轴正方向电场强度的增大而增大,分子极性减弱,趋于非极性分子;随着X轴正向电场增加能隙逐渐减小,分子总能量增加,分子变的不稳定,活性增加.
3)随着外电场强度的增加,第1、2、8、9激发能先增大后减小,第3、4、5、6、7激发能逐渐增加;第1、2、9激发态波长先减小后增大,第3、4、5、6、7、8激发态波长逐渐增大,这是因为外电场改变了CF3I分子轨道能级顺序和电子跃迁状态. 第1、2、4、5、6振子强度在0~0.009a.u.电场强度范围内为零,这表明第1、2、4、5、6激发态属于禁阻跃迁,第3、9振子强度随着电场强度的增加逐渐减小,激发的能力逐渐减小,第7、8振子强度随着电场的增强逐渐增大,激发的能力逐渐增强. 这为利用外电场控制CF3I分子的激发波长和强度提供了理论基础.
