X连锁高IgM综合征4例临床分析
2019-03-06李小青耿玲玲
张 翠,李小青 ,耿玲玲,冯 媛,南 楠,刘 昱
西安市儿童医院免疫科 (西安710003)
高IgM综合征(Hyper-IgM syndromes,HIGM)是一组少见的原发性免疫缺陷病(Primary immunodefieiency diseases,PID),发病机制主要为免疫球蛋白类别转换(Class switch recombination,CSR)障碍伴或不伴体细胞高频突变(Somatic hrper mutation,SHM)缺陷[1]。其主要特点为反复感染,血清IgG、IgA水平降低,IgM水平正常或升高。现已明确与HIGM发病相关的致病基因包括CD40L、AID、CD40、UNG、NEMO和IκBα基因[2]。其中CD40L基因位于Xq26,为X连锁隐性遗传,CD40L基因突变导致X连锁高IgM综合征(X-linked hyper-IgM syndromes,XHIGM),占所有HIGM病例数的65%~70%[3-4]。本组4例患者均为CD40L基因突变导致的X连锁高IgM综合征。
对象和方法
1 研究对象 2015年9月至2017年9月在本院就诊的,4例患儿均为男性,临床表现均为反复感染,中性粒细胞降低或缺乏,且经免疫球蛋白检测提示IgG显著降低,但IgM正常,T、B、NK淋巴细胞流式分析基本正常,临床高度怀疑高IgM综合征。
2 研究方法
2.1 抽取患儿及其父母静脉血各2ml,置入EDTA抗凝管中,联系基因公司,采用高通量基因测序技术对4例患儿进行基因检测,发现致病性突变后行一代测序进行验证,同时完成父母的基因测序,了解突变基因的来源。
2.2 收集4例患儿临床资料,包括发病年龄、确诊年龄、临床特征、实验室检查、基因突变位点以及治疗情况。
结果
1 基因诊断 经基因诊断,4例患儿为CD40L基因突变,诊断为X连锁高IgM综合征,3例母亲为携带者,1例为自发突变。基因检测结果见表1。
2 临床表现 4例XHIGM患儿平均起病年龄1.3岁,平均确诊年龄5.4岁,见表1。反复口腔溃疡平均起病年龄为2.3岁,现4例患儿均存活,其中1例患儿有一哥哥反复感染,并于10岁时死于化脓性脑膜炎,另3例患儿家族史阴性。确诊时收集到的临床主要表现为:反复口腔溃疡(4/4)、反复呼吸道感染(4/4)、化脓性中耳炎(2/4),化脓性扁桃体炎(1/4)、手足口病合并脑炎(1/4)、卡介苗病(1/4)、阑尾炎(1/4)、淋巴结炎(1/4),慢性浅表性胃炎(1/4)。
3 辅助检查 4例XHIGM患儿体液免疫均提示IgM正常或升高,IgG显著降低,2例IgA显著减低,2例IgA水平升高;T、B、NK淋巴细胞流式分析检测基本正常;白细胞正常或减低,中性粒细胞均减低,见表1。4例患儿进行骨髓检查结果分别示:骨髓粒系成熟减低,成熟延迟(3例);感染骨髓象(1例)。
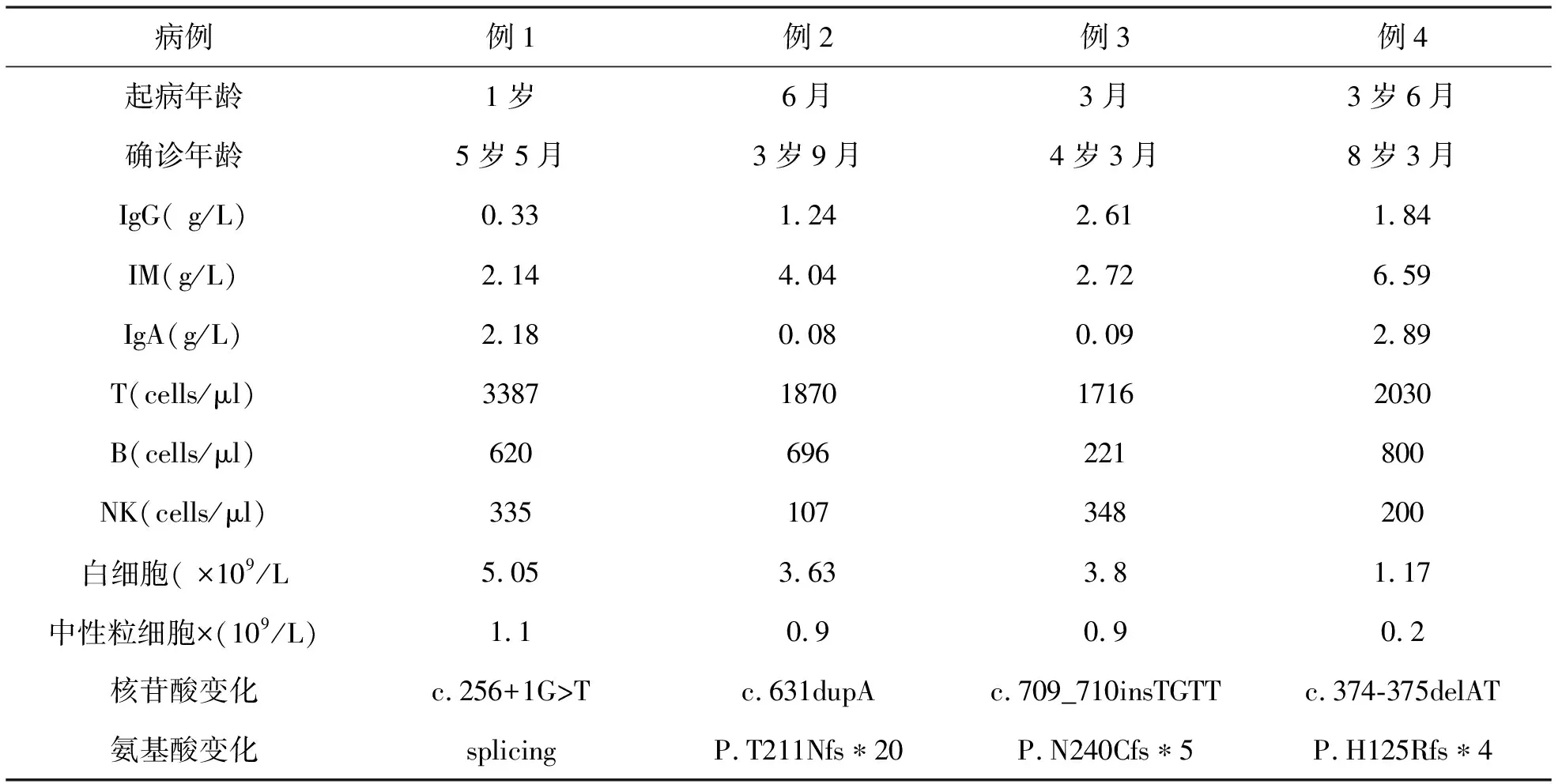
表1 4例患儿临床表现
4 治疗方法 4例患儿确诊XHIGM后给予静注入免疫球蛋白(IVIG)替代治疗,400~600mg/(kg·次),因患儿家长经济原因替代治疗间隔时间1月至半年。偶尔使用重组人粒细胞集落刺激因子升粒细胞。
讨论
X连锁高IgM综合征是一种原发性免疫缺陷病,在美国发病率预测为1∶1,000,000[5],国内目前尚无确切的发病率统计。XHIGM的致病基因为CD40L基因,CD40L是TNF超家族成员,主要表达于活化的CD4+T淋巴细胞表面,基因突变发生后可使T淋巴细胞表面CD40L表达缺如,损伤T淋巴细胞和B淋巴细胞相互作用,破坏生发中心形成,并影响CSR[3-4]。CD40L基因位于Xq26.3-27.1,为X连锁隐性遗传,约占HIGM的65%~70%。CD40L基因包含5个外显子,第l号外显子编码胞质区、跨膜区和胞外区6个氨基酸,第2号与第3号外显子编码胞外茎区,第4号、第5号外显子编码C末端147个氨基酸。据报道已发现160多种不同的基因突变,其中位于5号外显子者最为常见。本组4例病人中有2例患儿的突变发生在5号外显子,分别是例2和例3,与国外报道一致。这些突变或导致CD40L蛋白表达缺如或干扰CD40L三聚体形成及其与CD40结合,造成B细胞及T细胞活化障碍。
XHIGM典型的临床表现如下:反复感染、反复中性粒细胞减低以及家族中男性患病,血清IgG、IgA、IgE水平降低、IgM水平正常或升高,外周血B淋巴细胞计数正常[5-6]。根据典型的临床表现,对疑诊病人进行CD40L蛋白流式检测,如提示缺如,进而对CD40L基因测序发现致病突变明确诊断。但CD40L蛋白流式检测正常不能排除XHIGM,既往有文献报道2例CD40L蛋白流式检查结果正常的XHIGM患者[7-8],故当临床表现符合XHIGM时,即使CD40L蛋白流式检查结果正常,仍需进行CD40L基因检测。本组研究中2例患者IgA水平正常,与典型的XHIGM免疫学表现不同,在诊断初期,通过临床表现确定致病基因的环节,错误的将其疑诊为普通变异型免疫缺陷病,后经高通量测序技术查找到致病基因,故当根据临床表现不能确定致病基因,或根据怀疑的致病基因不能发现基因突变,可借助高通量测序技术(High throughput sequencing,HTS)查找致病基因。对于原发性免疫缺陷病目前临床上通过DNA测序技术,可以检测出大部分的点突变及部分小片段的插入缺失突变等。但由于原发性免疫缺陷病的遗传异质性和较大片段的基因插入或缺失,传统的Sanger测序技术已经远远不能满足临床应用及医学研究的需要。虽然近年来对PID疾病的认识在快速发展, 分子生物学和免疫学手段不断进步,但迄今在西方国家仍有约 1/3的PID患者无法明确致病基因。国内基因诊断不明的PID人数可能就更多,基因诊断几乎是PID确诊的唯一方法,但相当数量的PID 由于目前遗传学诊断技术的限制,还无法获得确定的诊断结果。高通量测序分析技术又称为二代测序技术(Next generation sequencing,NGS),采用基于HTS技术的多基因靶向测序技术可以将众多已知的原发性免疫缺陷病(Primary immunodefieiency diseases,PID)相关基因打包形成多基因并行测序项目,通过一次目前耗资约数千元的HTS,可获得所有被打包的候选PID致病基因检测结果。目前已有基因公司可以提供该项服务。本组病人即采用根据PID已知致病基因研发的靶向HTS服务,这些新技术的运用,已经使得部分疑难的PID病例得以确诊[9-10]。
XHIGM典型的免疫学表现:血清IgG、IgA水平降低、IgM水平正常或升高。在本组研究中,4例患者的IgM及IgG与典型的XHIGM临床表现相同,但不同的是本组2例患者IgA水平升高。有研究发现所有的患者均有显著的IgG 缺乏,大约50%的患者IgM水平升高,绝大多数患者有IgA缺乏,然而有一些患者IgA水平正常或升高[6]。国内也有少数XHIGM患儿IgA水平正常的报道[4]。因此IgA水平正常或升高不能排除XHIGM,提示IgA可由非经典途径产生,即IgA由肠道固有层B细胞在CpG及增殖诱导配体作用下活化产生[11]。
在过去的20年关于XHIGM的临床特征的回顾性分析已经被报道。在超过50%的病例中存在持续或间断的中性粒细胞减低。有报道显示68%的XHIGM 患者出现中性粒细胞减少,且粒细胞减少约45%为持续性的,大剂量IVIG对半数粒细胞减少患者有效,如果粒细胞减少严重,给予粒细胞集落刺激因子治疗有效[12]。本研究4例患者均有粒细胞减少,并伴有口腔溃疡,目前中性粒细胞减少原因尚未阐明,有研究显示可能与炎症微环境下CD40信号通路介导的粒细胞生成缺陷有关[13-14]。 已报道的疾病谱包括生后反复的细菌感染,机会性致病微生物如卡氏肺囊虫、隐孢子虫、弓形体、结核分支杆菌的易患性明显增加等。自身免疫性疾病及恶性肿瘤发病率明显升高,各种胃肠肿瘤、肝细胞癌、腺癌、胆管癌均可发生。细小病毒导致的红细胞减少症以及不明原因的渐进的神经退行性病变也有报导[ 15]。该病总体预后差,大约只有20%患者存活至25岁[16-18 ]。本组病人目前均存活,需继续长期随访,监测有无并发症,并及早干预。
目前骨髓干细胞移植(HCT)被认为是XHIGM唯一根治方法,此外其他治疗手段包括静注人免疫球蛋白替代治疗、预防性使用抗菌药物预防机会性感染、密切监测并发症例如中性粒细胞减低症和肝脏疾病、尽量避免隐孢子虫病暴露等。因为已报道的XHIGM患病率及死亡率显著高于正常人群,骨髓干细胞移植已经被认为是一个可能的根治该病的方法,关于骨髓干细胞移植成功的案例不断的积累,越来越多的患者接受了HCT[19-20]。Mt DLM等[21]总结了来自28个临床中心提供的189例XHIGM患者临床数据,时间跨度为1964至2013,其中176例患者进行了有效的随访并收集到有效的数据,其中67例患者接受了骨髓干细胞移植,随访平均时间为(8.5 ±7.2)年(0.1~36.2年),整体生存率在接受HCT的患者与未接受HCT的患者之间无统计学差异,而且肝脏疾病是影响总体生存率的重要因素。在存活者中,接受HCT的患者与未接受HCT的患者相比有更高的Lansky 和 Karnofsky 评分。在接受骨髓移植的患者中,27例出现了移植物抗宿主反应,大部分死亡发生在移植后1年内。所以接受骨髓移植的患者在一定程度上生活质量提高,但同时承受了由骨髓干细胞移植带来的更高的风险。Kanako Mitsui-Sekinaka等[22]报道日本56例XHIGM,其中29例患者接受了骨髓干细胞移植,另外27例未接受骨髓移植的患者在40岁时的长期存活率仅为28.2%。接受骨髓移植的患者的整体存活率显著高与未接受骨髓移植的患者,而且,≤5岁接受骨髓移植的患者的无病生存率显著高于>5岁接受骨髓移植的患者,Kanako Mitsui-Sekinaka等推断骨髓移植能显著改善患者的预后,认为≤5岁是接受骨髓移植的理想年龄,因为持续性的感染以及脏器损害经常在大于6岁的患者中发现。该组4例XHIGM患儿均未进行骨髓干细胞移植,仅给予静注入免疫球蛋白替代治疗。本组患者确诊年龄平均为5.4岁,超过了文献中提到的理想年龄,因此,对临床怀疑XHIGM的患儿尽早进行基因诊断,更有利与接受骨髓移植,改善预后。为了制定更理想的XHIGM管理方法,关于接受及未接受HCT的患者需要更多的最新随访结果,尤其是接受骨髓干细胞移植的患者的并发症需要关注。由于XHIGM致病基因已明确,使得基因治疗有望成为治愈XHIGM极有潜力的手段,但以病原微生物作为载体转人体内其致病性难以预料,故其安全性仍存在争议。
综上所述,当患儿反复感染、反复中性粒细胞减低以及家族中男性患病,血清IgG、IgA、IgE水平降低、IgM水平正常或升高,外周血B淋巴细胞计数正常,临床高度怀疑XHIGM可进行CD40L蛋白流式快速检测,当CD40L蛋白缺如时,提示诊断,进一步行CYBB基因测序,对于根据临床不能确定致病基因的患儿,可采用高通量测序技术,查找致病基因。对XHIGM患儿需早期基因检测,尽早明确诊断,早期干预,以期改善预后。
